

umwelt-online: Verordnung (EG) Nr. 152/2009 zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln (5)
|
zurück | |
| Analysemethoden zur Untersuchung von Futtermitteln auf das Vorhandensein nicht mehr zugelassener Zusatzstoffe | Anhang VIII |
| Wichtiger Hinweis:
Zum Nachweis des Vorhandenseins nicht mehr zugelassener Zusatzstoffe in Futtermitteln können empfindlichere Analysemethoden als die in diesem Anhang beschriebenen verwendet werden. Die in diesem Anhang aufgeführten Analysemethoden werden für Bestätigungszwecke herangezogen. |
A. Bestimmung des Methylbenzoquatgehalts
7-Benzyloxy-6-butyl-3-methoxycarbonyl-4-chinolon
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Methylbenzoquatgehalts von Futtermitteln. Die Bestimmungsgrenze beträgt 1 mg/kg.
2. Prinzip
Methylbenzoquat wird mit methanolischer Methansulfonsäure-Lösung aus der Probe extrahiert. Der Extrakt wird mit Dichlormethan, mittels Ionenaustauschchromatografie und anschließend erneut mit Dichlormethan gereinigt. Der Methylbenzoquatgehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (HPLC) unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
3.1 Dichlormethan.
3.2 Methanol, HPLC-Qualität.
3.3 Mobile Phase für die HPLC
Gemisch aus Methanol ( 3.2) und Wasser (HPLC-Qualität) 75 + 25 (V+V).
Die Lösung wird durch einen 0,22- µm-Filter ( 4.5) filtriert und entgast (z.B. durch 10-minütige Ultraschallbehandlung).
3.4 Methansulfonsäure-Lösung, c = 2 %
20,0 ml Methansulfonsäure werden mit Methanol ( 3.2) auf 1.000 ml verdünnt.
3.5 Salzsäure, c = 10 %
100 ml Salzsäure (ρ201,18 g/ml) werden mit Wasser auf 1.000 ml verdünnt.
3.6. Kationenaustauschharz Amberlit CG-120 (Na), 100 bis 200 Mesh
Dieses Harz wird vor der Verwendung vorbehandelt. Von dem Harz werden 100 g mit 500 ml verdünnter Salzsäure ( 3.5) aufgeschlämmt und auf einer Heizplatte unter ständigem Rühren zum Sieden gebracht. Es wird abkühlen gelassen, und die Säure wird dekantiert. Anschließend wird durch einen Papierfilter unter Vakuum filtriert. Das Harz wird 2-mal mit je 500 ml Wasser und danach mit 250 ml Methanol ( 3.2) gewaschen. Anschließend wird das Harz nochmals mit 250 ml Methanol gespült und der Filterkuchen trocken gesaugt. Das getrocknete Harz wird in einer verschlossenen Flasche aufbewahrt.
3.7 Standardsubstanz: Methylbenzoquat (7-Benzyloxy-6-butyl-3-methoxycarbonyl-4-chinolon), rein
3.7.1 Methylbenzoquat-Standard-Stammlösung , 500 µg/ml
Von der Standardsubstanz ( 3.7) werden 50 mg auf 0,1 mg genau eingewogen, in einem 100-ml-Messkolben in methanolischer Methansulfonsäure ( 3.4) gelöst, zur Marke aufgefüllt und gemischt.
3.7.2 Methylbenzoquat-Standardlösung, 50 µ g/ml
Von der Methylbenzoquat-Standard-Stammlösung ( 3.7.1) werden 5,0 ml in einen 50-ml-Messkolben überführt. Es wird mit Methanol ( 3.2) zur Marke aufgefüllt und gemischt.
3.7.3 Kalibrierlösungen
Von der Methylbenzoquat-Standardlösung ( 3.7.2) werden 1,0, 2,0, 3,0, 4,0 bzw. 5,0 ml jeweils in einen 25-ml-Messkolben überführt. Mit der mobilen Phase ( 3.3) wird zur Marke aufgefüllt und durchmischt. Diese Lösungen enthalten Methylbenzoquat in Konzentrationen von 2,0, 4,0, 6,0, 8,0 bzw. 10,0 µg/ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden.
4. Geräte
4.1 Laborschüttler.
4.2 Rotationsfilmverdampfer.
4.3 Glassäule (250 × 15 mm) mit Absperrhahn und Reservoir mit einem Fassungsvermögen von ca. 200 ml.
4.4 HPLC-Einrichtung mit UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor
4.4.1 HPLC-Trennsäule, 300 × 4 mm, C18, 10 µm Korngröße, oder vergleichbare Säule.
4.5 Membranfilter, 0,22m Porengröße.
4.6 Membranfilter, 0,45m Porengröße.
5. Verfahren
5.1 Allgemeines
5.1.1 Zur Prüfung, dass weder Methylbenzoquat noch Störsubstanzen vorhanden sind, wird eine Blindprobe untersucht.
5.1.2 Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Methylbenzoquat angereichert wurde. Die zugesetzte Menge an Methylbenzoquat sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 15 mg/kg werden 20 g der Blindprobe mit 600l Standard-Stammlösung ( 3.7.1) versetzt. Es wird gemischt und 10 min stehen gelassen, bevor mit der Extraktion ( 5.2) fortgefahren wird.
Für den Zweck dieser Methode muss die Blindprobe ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Methylbenzoquat darf nicht nachweisbar sein.
5.2 Extraktion
Von der vorbereiteten Probe werden 20 g auf 0,01 g genau in einen 250-ml-Erlenmeyerkolben eingewogen. Nach Zugabe von 100,0 ml Methansulfonsäure-Lösung ( 3.4) wird mechanisch ( 4.1) 30 min geschüttelt. Die Lösung wird durch ein Filterpapier filtriert und das Filtrat für die Flüssig-Flüssig-Trennung ( 5.3) aufbewahrt.
5.3 Flüssig-Flüssig-Trennung
Von dem durch 5.2 erhaltenen Filtrat werden 25,0 ml in einen 500-ml-Scheidetrichter überführt, der 100 ml Salzsäure ( 3.5) enthält. Nach Zugabe von 100 ml Dichlormethan ( 3.1) wird 1 min geschüttelt, die Phasentrennung abgewartet und die untere Phase (Dichlormethan) in einen 500-ml-Rundkolben ablaufen gelassen. Die Extraktion der wässrigen Phase wird 2-mal mit je 40 ml Dichlormethan wiederholt und die Extrakte werden zum ersten Extrakt im Rundkolben gegeben. Das Dichlormethanextrakt wird am Rotationsverdampfer ( 4.2) bei 40 °C und vermindertem Druck bis zur Trockne eingeengt. Der Rückstand wird in 20 bis 25 ml Methanol ( 3.2) gelöst und der Kolben verschlossen. Der gesamte Extrakt wird für die Ionenaustauschchromatografie ( 5.4) aufbewahrt.
5.4 Ionenaustauschromatografie
5.4.1 Vorbereitung der Kationenaustauschsäule
In das untere Ende der Glassäule ( 4.3) wird ein Glaswattebausch eingebracht. Von dem behandelten Kationenaustauschharz ( 3.6) werden 5,0 g mit 50 ml Salzsäure ( 3.5) aufgeschlämmt, in die Glassäule gegeben und absetzen gelassen. Nachdem der Säureüberschuss bis knapp zur Harzoberfläche abgelaufen ist, wird die Säule so lange mit Wasser gewaschen, bis die Waschflüssigkeit neutral gegen Lackmus ist. Dann werden 50 ml Methanol ( 3.2) auf die Säule gegeben und bis zur Harzoberfläche ablaufen gelassen.
5.4.2 Säulenchromatografie
Der Extrakt ( 5.3) wird mittels einer Pipette vorsichtig auf die Säule gegeben. Der Rundkolben wird 2-mal mit je 5 bis 10 ml Methanol ( 3.2) gespült. Die anfallenden Spüllösungen werden ebenfalls auf die Säule gegeben. Nachdem der Extrakt bis zur Harzoberfläche abgelaufen ist, wird die Säule mit 50 ml Methanol gewaschen, wobei die Flussrate höchstens 5 ml/min betragen darf. Die Waschflüssigkeit wird verworfen. Das Methylbenzoquat wird mit 150 ml methanolischer Methansulfonsäure-Lösung ( 3.4) von der Säule eluiert und das Eluat in einem 250-ml-Erlenmeyerkolben aufgefangen.
5.5 Flüssig-Flüssig-Trennung
Das Eluat ( 5.4.2) wird in einen 1-l-Scheidetrichter überführt. Der Erlenmeyerkolben wird mit 5 bis 10 ml Methanol ( 3.2) gespült und die anfallende Spülflüssigkeit ebenfalls in den Scheidetrichter gegeben. Dann werden 300 ml Salzsäure-Lösung (3.5) und 130 ml Dichlormethan ( 3.1) zugefügt. Es wird 1 min geschüttelt und die Phasentrennung abgewartet. Die untere Phase (Dichlormethan) wird in einen 500-ml-Rundkolben abgelassen. Die Extraktion der wässrigen Phase wird 2-mal mit je 70 ml Dichlormethan wiederholt, und diese Extrakte werden zum ersten Extrakt im Rundkolben gegeben.
Der Dichlormethanextrakt wird am Rotationsverdampfer ( 4.2) bei 40 °C und vermindertem Druck bis zur Trockne eingeengt. Der Rückstand im Rundkolben wird in ca. 5 ml Methanol ( 3.2) gelöst und die Lösung quantitativ in einen 10-ml-Messkolben überführt. Der Rundkolben wird 2-mal mit je 1 bis 2 ml Methanol gespült, und die Spüllösungen werden in den Messkolben überführt. Es wird mit Methanol zur Marke aufgefüllt und durchmischt. Ein aliquoter Teil wird durch einen Membranfilter ( 4.6) filtriert. Die Lösung wird für die HPLCBestimmung ( 5.6) aufbewahrt.
5.6 HPLC-Bestimmung
5.6.1 Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung ( 3.7.3), die 4 µg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.6.2 Erstellung der Kalibrationskurve
Jede Kalibrierlösung ( 3.7.3) wird mehrmals eingespritzt, und die Peakhöhen (-flächen) für die einzelnen Konzentrationen werden gemessen. Es wird eine Kalibrationskurve gezeichnet, indem die mittleren Peakhöhen oder -flächen auf der Ordinate und die dazugehörigen Konzentrationen ing/ml auf der Abszisse aufgetragen werden.
5.6.3 Bestimmung der Probenlösung
Der Probenextrakt ( 5.5) wird mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird, und die mittlere Peakhöhe (-fläche) der Methylbenzoquatpeaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Methylbenzoquatpeaks der Probenlösung wird anhand der Kalibrationskurve ( 5.6.2) die Konzentration der Probenlösung in µg/ml bestimmt.
Der Methylbenzoquatgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
| w = | c x 40 |
| m |
wobei:
c = Methylbenzoquatkonzentration der Probenlösung in µg/ml.
m = Probeneinwaage in g.
7. Überprüfung der Ergebnisse
7.1 Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung und der Kalibrierlösung ( 3.7.3), die 10 µg/ml enthält, verglichen werden.
7.1.1 Co-Chromatografie
Ein Probenextrakt wird mit einer geeigneten Menge Methylbenzoquat-Standardlösung ( 3.7.2) versetzt. Die Menge des zugesetzten Methylbenzoquats muss dem erwarteten Methylbenzoquatgehalt des Probenextrakts entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Methylbenzoquatpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks der nicht angereicherten Probenlösung abweichen.
7.1.2 Diodenarray-Detektion
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf bei Methylbenzoquatgehalten von 4-20 mg/kg 10 % des höheren Wertes nicht überschreiten.
7.3 Wiederfindungsrate
Für eine angereicherte Blindprobe muss die Wiederfindungsrate mindestens 90 % betragen.
8. Ergebnisse eines Ringversuchs
Es wurden 5 Proben von 10 Laboratorien untersucht. Jede Probe wurde doppelt analysiert.
| Blindprobe | Mehl 1 | Pellets 1 | Mehl 2 | Pellets 2 | |
| Mittelwert [mg/kg] | NN | 4,50 | 4,50 | 8,90 | 8,70 |
| sr[mg/kg] | - | 0,30 | 0,20 | 0,60 | 0,50 |
| VKr[ %] | - | 6,70 | 4,40 | 6,70 | 5,70 |
| sR[mg/kg] | - | 0,40 | 0,50 | 0,90 | 1,00 |
| VKR[ %] | - | 8,90 | 11,10 | 10,10 | 11,50 |
| Wiederfindung [ %] | - | 92,00 | 93,00 | 92,00 | 89,00 |
NN = Nicht nachweisbar
sr = Standardabweichung der Wiederholbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
B. Bestimmung des Olaquindoxgehalts
N-(2-Hydroxyethyl)-3-methyl-2-chinoxalincarbamid-1,4-dioxid
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Olaquindoxgehalts von Futtermitteln. Die Bestimmungsgrenze beträgt 5 mg/kg.
2. Prinzip
Die Probe wird mit einem Methanol-Wasser-Gemisch extrahiert. Der Gehalt an Olaquindox wird durch Umkehrphasen-Hochleistungsflüssigkeitschromatografie (RP-HPLC) unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
3.1 Methanol.
3.2 Methanol, HPLC-Qualität.
3.3 Wasser, HPLC-Qualität.
3.4 Mobile Phase für die HPLC:
Wasser (3.3)-Methanol (3.2)-Gemisch, z.B. 900 + 100 (V+V).
3.5 Standardsubstanz: Olaquindox, rein, N-(2-Hydroxyethyl)-3-methyl-2-chinoxalincarbamid-1,4-dioxid, E 851
3.5.1 Olaquindox-Standard-Stammlösung, 250 µ g/ml
Von Olaquindox ( 3.5) werden 50 mg auf 0,1 mg genau in einen 200-ml-Messkolben eingewogen, und es werden 190 ml Wasser zugefügt. Dann wird der Kolben 20 min in ein Ultraschallbad ( 4.1) gestellt. Nach der Ultraschallbehandlung wird die Lösung auf Raumtemperatur gebracht, es wird mit Wasser zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt und im Kühlschrank aufbewahrt. Die Lösung muss monatlich frisch hergestellt werden.
3.5.2 Olaquindox-Standardlösung, 25 µg/ml
Von der Standard-Stammlösung ( 3.5.1) werden 10,0 ml in einen 100-ml-Messkolben überführt. Es wird mit der mobilen Phase (3.4) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt und im Kühlschrank aufbewahrt. Die Lösung muss täglich frisch hergestellt werden.
3.5.3 Kalibrierlösungen
Von der Standardlösung ( 3.5.2) werden 1,0, 2,0, 5,0, 10,0, 15,0 bzw. 20,0 ml jeweils in einen 50-ml-Messkolben überführt. Mit der mobilen Phase ( 3.4) wird zur Marke aufgefüllt und durchmischt. Der Kolben wird mit Aluminiumfolie umhüllt. Diese Kalibrierlösungen enthalten jeweils 0,5, 1,0, 2,5, 5,0, 7,5 bzw. 10,0 µg Olaquindox je ml.
Die Lösungen müssen vor Gebrauch frisch hergestellt werden.
4. Geräte
4.1 Ultraschallbad.
4.2 Mechanisches Schüttelgerät.
4.3 HPLC-Einrichtung mit UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor
4.3.1 HPLC-Trennsäule, 250 × 4 mm, C18, 10 µm Korngröße, oder vergleichbare Säule.
4.4 Membranfilter, 0,45 µm Porengröße.
5. Verfahren
Anmerkung:
Olaquindox ist lichtempfindlich. Alle Analysenschritte sind deshalb bei gedämpftem Licht oder unter Verwendung von Braunglasgeräten durchzuführen.
5.1 Allgemeines
5.1.1 Zur Prüfung, dass weder Olaquindox noch Störsubstanzen vorhanden sind, wird eine Blindprobe untersucht.
5.1.2 Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Olaquindox angereichert wurde. Die zugesetzte Menge an Olaquindox sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 50 mg/kg werden 10,0 ml der Standard-Stammlösung ( 3.5.1) in einen 250-ml-Erlenmeyerkolben überführt und im Stickstoffstrom auf ca. 0,5 ml eingeengt. Dann werden 50 g der Blindprobe zugegeben. Es wird gründlich gemischt und 10 min stehen gelassen, bevor mit der Extraktion ( 5.2) fortgefahren wird.
Anmerkung:
Für den Zweck dieser Methode muss die Blindprobe ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Olaquindox darf nicht nachweisbar sein.
5.2 Extraktion
Von der Probe werden 50 g auf 0,01 g genau in einen 1.000-ml-Erlenmeyerkolben eingewogen und mit 100 ml Methanol ( 3.1) versetzt. Der Kolben wird 5 min in das Ultraschallbad ( 4.1) gestellt. Es werden 410 ml Wasser zugegeben, und die Probenlösung wird weitere 15 min mit Ultraschall behandelt. Danach wird der Kolben aus dem Ultraschallbad genommen, und es wird 30 min auf dem Schüttelgerät ( 4.2) geschüttelt. Die Probenlösung wird durch einen Faltenfilter filtriert. Von dem Filtrat werden 10,0 ml in einen 20-ml-Messkolben überführt, es wird mit Wasser zur Marke aufgefüllt und gemischt. Ein aliquoter Teil wird durch einen Membranfilter ( 4.4) filtriert (siehe Bemerkung 9). Anschließend wird die HPLC-Bestimmung ( 5.3) durchgeführt.
5.3 HPLC-Bestimmung
5.3.1 Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
| Analysensäule ( 4.3.1) | |
| Mobile Phase ( 3.4): | Wasser ( 3.3)-Methanol ( 3.2)- Gemisch 900 + 100 (V+V) |
| Durchflussrate: | 1,5 bis 2 ml/min |
| Detektionswellenlänge: | 380 nm |
| Einspritzvolumen: | 20 bis 100 µl |
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung ( 3.5.3), die 2,5 µ g/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.3.2 Erstellung der Kalibrationskurve
Jede Kalibrierlösung ( 3.5.3) wird mehrmals eingespritzt, und die Peakhöhen (-flächen) für die einzelnen Konzentrationen werden gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen (-flächen) auf der Ordinate und die dazugehörigen Konzentrationen ing/ml auf der Abszisse aufgetragen werden.
5.3.3 Bestimmung der Probenlösung
Der Probenextrakt ( 5.2) wird mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird. Die mittlere Peakhöhe (-fläche) der Olaquindoxpeaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Olaquindoxpeaks der Probenlösung wird anhand der Kalibrationskurve ( 5.3.2) die Konzentration der Probenlösung in µg/ml bestimmt.
Der Olaquindoxgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
| w = | c x 1.000 |
| m |
wobei:
c = Olaquindoxkonzentration des Probenextrakts ( 5.2) in µg/ml.
m = Probeneinwaage in g.
7. Überprüfung der Ergebnisse
7.1 Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung ( 5.2) und der Kalibrierlösung ( 3.5.3), die 5,0g/ml enthält, verglichen werden.
7.1.1 Co-Chromatografie
Ein Probenextrakt ( 5.2) wird mit einer geeigneten Menge Kalibrierlösung ( 3.5.3) versetzt. Die zugesetzte Olaquindoxmenge muss dem erwarteten Olaquindoxgehalt des Probenextrakts entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Olaquindoxpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks des nicht angereicherten Probenextrakts abweichen.
7.1.2 Diodenarray-Detektion
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2 Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf bei einem Olaquindoxgehalt zwischen 10 und 200 mg/kg 15 % des höheren Wertes nicht überschreiten.
7.3 Wiederfindungsrate
Für eine angereicherte Blindprobe muss die Wiederfindungsrate mindestens 90 % betragen.
8. Ergebnisse eines Ringversuchs
Es wurde ein EG-Ringversuch durchgeführt, bei dem 4 Ferkelfuttermittel einschließlich eines Blindfuttermittels von bis zu 13 Laboratorien untersucht wurden. Die Ergebnisse sind in der nachstehenden Tabelle zusammengefasst.
| Probe 1 | Probe 2 | Probe 3 | Probe 4 | |
| L | 13 | 10 | 11 | 11 |
| n | 40 | 40 | 44 | 44 |
| Mittelwert [mg/kg] | - | 14,6 | 48,0 | 95,4 |
| sr[mg/kg] | - | 0,82 | 2,05 | 6,36 |
| sR[mg/kg] | - | 1,62 | 4,28 | 8,42 |
| VKr[ %] | - | 5,6 | 4,3 | 6,7 |
| VKR[ %] | - | 11,1 | 8,9 | 8,8 |
| Sollgehalt [mg/kg] |
- | 15 | 50 | 100 |
| Wiederfindung [ %] | - | 97,3 | 96,0 | 95,4 |
L = Anzahl der Laboratorien
n = Anzahl der Einzelwerte
sr = Standardabweichung der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
9. Bemerkung
Obwohl die Methode für Futtermittel mit Olaquindoxgehalten von mehr als 100 mg/kg nicht validiert wurde, können durch Verringerung der Einwaage und/oder Verdünnung des Extrakts ( 5.2) auf den Konzentrationsbereich der Kalibrationskurve ( 5.3.2) zufriedenstellende Ergebnisse erzielt werden.
C. Bestimmung des Amproliumgehalts
1-[(4-Amino-2-propylpyrimidin-5-yl)methyl]-2-methylpyridiniumchlorid-hydrochlorid
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Amproliumgehalts von Futtermitteln und Vormischungen. Die Nachweisgrenze beträgt 1 mg/kg, die Bestimmungsgrenze 5 mg/kg.
2. Prinzip
Die Probe wird mit einem Methanol-Wasser-Gemisch extrahiert. Nach Verdünnung mit der mobilen Phase und Membranfiltration wird der Amproliumgehalt mittels Hochleistungsflüssigkeitschromatografie (HPLC) über ein Kationenaustauschermaterial unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
3.1 Methanol.
3.2 Acetonitril, HPLC-Qualität.
3.3 Wasser, HPLC-Qualität.
3.4 Natriumdihydrogenphosphatlösung, c = 0,1 mol/l
Von dem Natriumdihydrogenphosphatmonohydrat werden 13,80 g in einem 1.000-ml-Messkolben in Wasser ( 3.3) gelöst. Es wird mit Wasser ( 3.3) zur Marke aufgefüllt und gemischt.
3.5 Natriumperchloratlösung, c = 1,6 mol/l
Von dem Natriumperchloratmonohydrat werden 224,74 g in einem 1.000-ml-Messkolben in Wasser ( 3.3) gelöst. Es wird mit Wasser ( 3.3) zur Marke aufgefüllt und gemischt.
3.6 Mobile Phase für die HPLC (siehe Bemerkung 9.1)
Mischung aus Acetonitril ( 3.2), Natriumdihydrogenphosphatlösung ( 3.4) und Natriumperchloratlösung ( 3.5), 450 + 450 + 100 (V+V+V). Vor der Verwendung wird die Lösung durch einen 0,22-µ m-Membranfilter ( 4.3) filtriert und entgast (z.B. mindestens 15 min im Ultraschallbad (4.4)).
3.7 Standardsubstanz: Amprolium, rein, 1-[(4-Amino-2-propylpyrimidin-5-yl)methyl]-2-methylpyridiniumchlorid-hydrochlorid, E 750 (siehe 9.2)
3.7.1 Amprolium-Standard-Stammlösung, 500 µg/ml
Von Amprolium ( 3.7) werden 50 mg auf 0,1 mg genau in einen 100-ml-Messkolben eingewogen, in 80 ml Methanol ( 3.1) gelöst und 10 min in ein Ultraschallbad ( 4.4) gestellt. Nach der Ultraschallbehandlung wird die Lösung auf Raumtemperatur gebracht; es wird mit Wasser zur Marke aufgefüllt und gemischt. Bei einer Temperatur von ≤ 4 °C ist diese Lösung 1 Monat lang haltbar.
3.7.2 Amprolium - Standardlösung, 50 µg/ml
Von der Standard-Stammlösung ( 3.7.1) werden 5,0 ml in einen 50-ml-Messkolben pipettiert; es wird mit der Extraktionslösung (3.8) zur Marke aufgefüllt und gemischt. Bei einer Temperatur von ≤4 °C ist diese Lösung 1 Monat lang haltbar.
3.7.3 Kalibrierlösungen
Von der Amprolium-Standardlösung ( 3.7.2) werden 0,5, 1,0 bzw. 2,0 ml jeweils in einen 50-ml-Messkolben überführt. Mit der mobilen Phase ( 3.6) wird zur Marke aufgefüllt und durchmischt. Diese Lösungen enthalten jeweils 0,5, 1,0 bzw. 2,0 µg Amprolium je ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden.
3.8 Extraktionslösung
Methanol ( 3.1)-Wasser-Mischung 2 + 1 (V+V).
4. Geräte
4.1 HPLC-Einrichtung mit Injektionssystem für Einspritzvolumina von 100 µl
4.1.1 HPLC-Trennsäule für die Kationenaustauschchromatografie, 125 × 4 mm, z.B. Nucleosil 10 SA, 5 oder 10 µm Korngröße, oder vergleichbare Säule.
4.1.2 UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor.
4.2 Membranfilter, PTFE-Material, 0,45 µm Porengröße.
4.3 Membranfilter, 0,22 µm Porengröße.
4.4 Ultraschallbad.
4.5 Mechanisches Schüttelgerät oder Magnetrührer.
5. Verfahren
5.1 Allgemeines
5.1.1 Blindprobe
Für die Durchführung des Wiederfindungstests ( 5.1.2) muss zur Prüfung, dass weder Amprolium noch Störsubstanzen vorhanden sind, eine Blindprobe untersucht werden. Die Blindprobe muss ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Amprolium oder Störsubstanzen dürfen nicht nachweisbar sein.
5.1.2 Wiederfindungstest
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Amprolium angereichert wurde. Die zugesetzte Menge an Amprolium sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 100 mg/kg werden 10,0 ml der Standard-Stammlösung ( 3.7.1) in einen 250-ml-Erlenmeyerkolben überführt und auf ca. 0,5 ml eingeengt. Dann werden 50 g der Blindprobe zugegeben. Es wird gründlich gemischt, 10 min stehen gelassen und nochmals mehrfach gemischt, bevor mit der Extraktion ( 5.2) fortgefahren wird.
Ist eine der zu untersuchenden Probe ähnliche Blindprobe nicht verfügbar (siehe 5.1.1), so kann ein Wiederfindungstest mithilfe des Additionsverfahrens durchgeführt werden. In diesem Fall wird die zu untersuchende Probe mit einer Amproliummenge angereichert, die der bereits in der Probe vorhandenen Menge entspricht. Diese Probe wird zusammen mit der nicht angereicherten Probe untersucht, und die Wiederfindungsrate kann durch Subtraktion ermittelt werden.
5.2 Extraktion
5.2.1 Vormischungen (Amproliumgehalt < 1 %) und Futtermittel
Je nach Amproliumgehalt werden 5 bis 40 g der Probe auf 0,01 g genau in einen 500-ml-Erlenmeyerkolben eingewogen und mit 200 ml Extraktionslösung ( 3.8) versetzt. Der Kolben wird 15 min in ein Ultraschallbad ( 4.4) gestellt und anschließend 60 min auf dem Schüttelgerät geschüttelt oder auf dem Magnetrührer ( 4.5) gerührt. Ein aliquoter Teil des Extrakts wird mit der mobilen Phase ( 3.6) auf einen Amproliumgehalt von 0,5 bis 2 µg/ml verdünnt und gemischt (siehe Bemerkung 9.3). Von dieser verdünnten Lösung werden 5 bis 10 ml durch einen Membranfilter ( 4.2) filtriert. Anschließend wird die HPLC-Bestimmung ( 5.3) durchgeführt.
5.2.2 Vormischungen (Amproliumgehalt ≥ 1 %)
Je nach Amproliumgehalt werden 1 bis 4 g der Vormischung auf 0,001 g genau in einen 500-ml-Erlenmeyerkolben eingewogen und mit 200 ml Extraktionslösung ( 3.8) versetzt. Der Kolben wird 15 min in ein Ultraschallbad ( 4.4) gestellt und anschließend 1 h auf dem Schüttelgerät geschüttelt oder auf dem Magnetrührer gerührt ( 4.5). Ein aliquoter Teil des Extrakts wird mit der mobilen Phase ( 3.6) auf einen Amproliumgehalt von 0,5 bis 2 µg/ml verdünnt und gemischt. Von dieser verdünnten Lösung werden 5 bis 10 ml durch einen Membranfilter ( 4.2) filtriert. Anschließend wird die HPLC-Bestimmung ( 5.3) durchgeführt.
5.3 HPLC-Bestimmung
5.3.1 Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren
| Ergebnissen führen: HPLC-Trennsäule ( 4.1.1): |
125 × 4 mm, Kationenaustausch Nucleosil 10 SA, 5 oder 10 pm Korngröße, oder vergleichbare Säule |
| Mobile Phase ( 3.6): | Mischung aus Acetonitril ( 3.2), Natriumdihydrogenphosphat-Lösung ( 3.4) und Natriumperchlorat-Lösung ( 3.5), 450 + 450 + 100 (V+V+V) |
| Durchflussrate: | 0,7 bis 1 ml/min |
| Detektionswellenlänge: | 264 nm |
| Einspritzvolumen: | 100 µl |
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung ( 3.7.3), die 1,0 }tg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.3.2 Erstellung der Kalibrationskurve
Jede Kalibrierlösung ( 3.7.3) wird mehrmals eingespritzt, und die mittleren Peakhöhen (-flächen) werden für die einzelnen Konzentrationen gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen (-flächen) auf der Ordinate und die dazugehörigen Konzentrationen in gg/ml auf der Abszisse aufgetragen werden.
5.3.3 Bestimmung der Probenlösung
Der Probenextrakt ( 5.2) wird mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird. Die mittlere Peakhöhe (-fläche) der Amproliumpeaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Amproliumpeaks der Probenlösung wird anhand der Kalibrationskurve ( 5.3.2) die Konzentration der Probenlösung in gg/ml bestimmt.
Der Amproliumgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
| w = | V x c x f | [mg/kg] |
| m |
wobei:
V = Volumen der Extraktionslösung ( 3.8) in ml gemäß 5.2 (d. h. 200 ml),
c = Amproliumkonzentration der Probenlösung ( 5.2) in µg/ml,
f = Verdünnungsfaktor gemäß 5.2,
m = Probeneinwaage in g.
7. Überprüfung der Ergebnisse
7.1 Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung ( 5.2) und der Kalibrierlösung ( 3.7.3), die 2,0 µg/ml enthält, verglichen werden.
7.1.1 Co-Chromatografie
Eine Probenlösung ( 5.2) wird mit einer geeigneten Menge Kalibrierlösung ( 3.7.3) angereichert. Die zugesetzte Amproliummenge muss dem Amproliumgehalt des Probenextrakts entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Amproliumpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks der nicht angereicherten Probenlösung abweichen.
7.1.2 Diodenarray-Detektion
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
Wird eines dieser Kriterien nicht erfüllt, so gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2 Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
7.3. Wiederfindungsrate
Bei einer angereicherten (Blind-)Probe muss die Wiederfindungsrate mindestens 90 % betragen.
8. Ergebnisse eines Ringversuchs
Bei einem Ringversuch wurden 3 Geflügelfuttermittel (Proben 1 bis 3), 1 Mineralfuttermittel (Probe 4) und 1 Vormischung (Probe 5) untersucht. Die Ergebnisse sind in der nachstehenden Tabelle zusammengefasst.
| Probe 1 (Blindprobe) |
Probe 2 | Probe 3 | Probe 4 | Probe 5 | |
| L | 14 | 14 | 14 | 14 | 15 |
| n | 56 | 56 | 56 | 56 | 60 |
| Mittelwert [mg/kg] | - | 45,5 | 188 | 5 129 | 25 140 |
| sr[mg/kg] | - | 2,26 | 3,57 | 178 | 550 |
| VKr[ %] | - | 4,95 | 1,90 | 3,46 | 2,20 |
| sR[mg/kg] | - | 2,95 | 11,8 | 266 | 760 |
| VKR[ %] | - | 6,47 | 6,27 | 5,19 | 3,00 |
| Sollgehalt [mg/kg] | - | 50 | 200 | 5 000 | 25 000 |
L = Anzahl der Laboratorien
n = Anzahl der Einzelwerte
sr = Standardabweichung der Wiederholbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
9. Bemerkungen
9.1 Enthält die Probe Thiamin, so erscheint der Thiaminpeak in der Chromatografie kurz vor dem Amproliumpeak. Bei Anwendung dieser Methode müssen Amprolium und Thiamin getrennt werden. Sind Amprolium und Thiamin nicht durch die bei dieser Methode verwendete Säule ( 4.1.1) getrennt, sollten bis zu 50 % des Acetonitrilanteils der mobilen Phase ( 3.6) durch Methanol ersetzt werden.
9.2 Gemäß dem britischen Arzneibuch zeigt das Spektrum einer Amproliumlösung (c = 0,02 mol/l) in Salzsäure (c = 0,1 mol/l) Maxima bei 246 nm und 262 nm. Die Absorption beträgt 0,84 bei 246 nm bzw. 0,80 bei 262 nm.
9.3 Der Extrakt muss immer mit der mobilen Phase verdünnt werden, da sich sonst die Retentionszeit des Amproliumpeaks durch Veränderungen der Ionenstärke beträchtlich verschieben kann.
D. Bestimmung des Carbadoxgehalts
Methyl 3-(2-chinoxalinylmethylen)carbazat N1,N4-dioxid
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Carbadoxgehalts von Futtermitteln, Vormischungen und Zubereitungen. Die Nachweisgrenze beträgt 1 mg/kg, die Bestimmungsgrenze 5 mg/kg.
2. Prinzip
Die Probe wird mit Wasser äquilibriert und anschließend mit Methanol-Acetonitril extrahiert. Bei Futtermitteln wird ein aliquoter Teil des filtrierten Extrakts auf einer Aluminiumoxid-Säule gereinigt. Bei Vormischungen und Zubereitungen wird ein aliquoter Teil des filtrierten Extrakts mit einem Gemisch aus Wasser, Methanol und Acetonitril auf eine geeignete Konzentration verdünnt. Der Carbadoxgehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (HPLC) unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
3.1 Methanol.
3.2 Acetonitril, HPLC-Qualität.
3.3 Essigsäure, w = 100 %.
3.4 Aluminiumoxid: neutral, Aktivitätsstufe I.
3.5 Methanol-Acetonitril 1 + 1 (V+V)
500 ml Methanol ( 3.1) werden mit 500 ml Acetonitril ( 3.2) gemischt.
3.6 Essigsäure, σ = 10 %
10 ml Essigsäure ( 3.3) werden auf 100 ml in Wasser gelöst.
3.7 Natriumacetat.
3.8 Wasser, HPLC-Qualität.
3.9 Acetat-Pufferlösung, c = 0,01 mol/l, pH = 6,0
0,82 g Natriumacetat ( 3.7) werden in 700 ml Wasser ( 3.8) gelöst, und der pH-Wert wird mit Essigsäure ( 3.6) auf 6,0 eingestellt. Die Lösung wird in einen 1 000-ml-Messkolben überführt. Es wird mit Wasser ( 3.8) zur Marke aufgefüllt und gemischt.
3.10 Mobile Phase für die HPLC
825 ml der Acetat-Pufferlösung ( 3.9) werden mit 175 ml Acetonitril ( 3.2) vermischt.
Die Lösung wird durch einen 0,22-µm-Filter ( 4.5) filtriert und entgast (z.B. durch 10-minütige Ultraschallbehandlung);
3.11 Standardsubstanz
Carbadox, rein: Methyl 3-(2-chinoxalinylmethylen)carbazat N1,N4-dioxid, E 850
3.11.1 Carbadox-Standard-Stammlösung, 100 µg/ml (siehe Anmerkung unter " 5. Verfahren")
Von der Carbadox-Standardsubstanz ( 3.11) werden 25 mg auf 0,1 mg genau in einen 250-ml-Messkolben eingewogen und im Ultraschallbad (4.7) in Methanol-Acetonitril ( 3.5) gelöst. Nach der Ultraschallbehandlung wird die Lösung auf Raumtemperatur gebracht, zur Marke mit Methanol-Acetonitril ( 3.5) aufgefüllt und gemischt. Der Kolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 °C ist diese Lösung 1 Monat lang haltbar.
3.11.2 Kalibrierlösungen
Von der Standard-Stammlösung ( 3.11.1) werden 2,0, 5,0, 10,0 bzw. 20,0 ml jeweils in einen 100-ml-Messkolben überführt. Es werden jeweils 30 ml Wasser hinzugefügt; es wird mit Methanol-Acetonitril ( 3.5) zur Marke aufgefüllt und gemischt. Die Kolben werden mit Aluminiumfolie umhüllt. Diese Lösungen enthalten jeweils 2,0, 5,0, 10,0 bzw. 20,0 µg/ml Carbadox.
Kalibrierlösungen müssen vor Gebrauch frisch hergestellt werden.
Anmerkung:
Für die Bestimmung des Carbadoxgehalts in Futtermitteln, die weniger als 10 mg/kg enthalten, müssen Kalibrierlösungen mit einer Konzentration von weniger als 2,0 µg/ml bereitet werden.
3.12. Wasser-[Methanol-Acetonitril ( 3.5)]-Gemisch, z.B. 300 + 700 (V+V).
300 ml Wasser werden mit 700 ml des Methanol-Acetonitril-Gemisches ( 3.5) vermischt.
4. Geräte
4.1 Laborschüttler oder Magnetrührer.
4.2 Glasfaser-Filterpapier, z.B. Whatman GF/a oder gleichwertig.
4.3 Glassäule (Länge 300 bis 400 mm, Innendurchmesser etwa 10 mm) mit Sinterglasfritte und Auslaufhahn
Anmerkung:
Eine Glassäule mit Absperrhahn oder eine konisch zulaufende Glassäule können ebenfalls verwendet werden; in diesem Fall wird ein kleiner Glaswattebausch in den unteren Teil der Säule eingebracht und mit einem Glasstab zusammengedrückt.
4.4 HPLC-Einrichtung mit Injektionssystem für Einspritzvolumina von 20l
4.4.1 HPLC-Trennsäule, 300 × 4 mm, C18, 10m Korngröße, oder vergleichbare Säule.
4.4.2 UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor mit einem Messbereich von 225 bis 400 nm.
4.5 Membranfilter, 0,22 µm Porengröße.
4.6 Membranfilter, 0,45 µm Porengröße.
4.7 Ultraschallbad.
5. Verfahren
Anmerkung:
Carbadox ist lichtempfindlich. Alle Analysenschritte sind daher bei gedämpftem Licht oder unter Verwendung von Braunglasgeräten durchzuführen. Alternativ können die Glaswaren mit Aluminiumfolie umwickelt werden.
5.1 Allgemeines
5.1.1 Blindprobe
Für die Durchführung des Wiederfindungstests ( 5.1.2) muss zur Prüfung, dass weder Carbadox noch Störsubstanzen vorhanden sind, eine Blindprobe untersucht werden. Die Blindprobe muss ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Carbadox oder Störsubstanzen dürfen nicht nachweisbar sein.
5.1.2 Wiederfindungstest
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe ( 5.1.1) untersucht wird, die mit Carbadox angereichert wurde. Die zugesetzte Menge an Carbadox sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 50 mg/kg werden 5,0 ml der Standard-Stammlösung ( 3.11.1) in einen 200-ml-Erlenmeyerkolben gegeben. Die Lösung wird in einem Stickstoffstrom auf ca. 0,5 ml eingeengt. Dann werden 10 g der Blindprobe zugegeben. Es wird gemischt und 10 min stehen gelassen, bevor mit dem Extraktionsschritt ( 5.2) fortgefahren wird.
Ist eine der zu untersuchenden Probe ähnliche Blindprobe nicht verfügbar (siehe 5.1.1), so kann ein Wiederfindungstest mithilfe des Standard-Additionsverfahrens durchgeführt werden. In diesem Fall wird die zu untersuchende Probe mit einer Carbadoxmenge angereichert, die der bereits in der Probe vorhandenen Menge entspricht. Diese Probe wird zusammen mit der nicht angereicherten Probe untersucht, und die Wiederfindungsrate kann durch Subtraktion ermittelt werden.
5.2 Extraktion
5.2.1 Futtermittel
Von der Probe werden 10 g auf 0,01 g genau in einen 200-ml-Erlenmeyerkolben eingewogen. Nach Zugabe von 15,0 ml Wasser wird gemischt und 5 min stehen gelassen. Dann werden 35,0 ml Methanol-Acetonitril ( 3.5) hinzugefügt, der Kolben wird verschlossen und 30 min auf dem Schüttelgerät geschüttelt oder auf dem Magnetrührer ( 4.1) gerührt. Die Lösung wird über Glasfaser-Filterpapier ( 4.2) filtriert. Diese Lösung wird für die Reinigung (5.3) aufbewahrt.
5.2.2 Vormischungen (0,1 bis 2,0 % )
Von der nicht gemahlenen Probe wird 1 g auf 0,001 g genau in einen 200-ml-Erlenmeyerkolben eingewogen. Nach Zugabe von 15,0 ml Wasser wird gemischt und 5 min stehen gelassen. Dann werden 35,0 ml Methanol-Acetonitril ( 3.5) hinzugefügt, der Kolben wird verschlossen und 30 min auf dem Schüttelgerät geschüttelt oder auf dem Magnetrührer ( 4.1) gerührt. Die Lösung wird über Glasfaser-Filterpapier ( 4.2) filtriert.
Ein aliquoter Teil des Filtrats wird in einen 50-ml-Messkolben pipettiert. Es werden 15,0 ml Wasser hinzugefügt; es wird zur Marke mit Methanol-Acetonitril ( 3.5) aufgefüllt und gemischt. Die Carbadox-Konzentration der fertigen Lösung muss bei etwa 10 µg/ml liegen. Ein aliquoter Teil wird durch einen 0,45-µm-Membranfilter ( 4.6) filtriert.
Anschließend wird die HPLC-Bestimmung ( 5.4) durchgeführt.
5.2.3 Zubereitungen (> 2 % )
Von der nicht gemahlenen Probe werden 0,2 g auf 0,001 g genau in einen 250-ml-Erlenmeyerkolben eingewogen. Es werden 45,0 ml Wasser hinzugefügt, gemischt und 5 min stehen gelassen. Dann werden 105,0 ml Methanol-Acetonitril ( 3.5) hinzugefügt. Der Kolben wird verschlossen, geschüttelt, 15 min in ein Ultraschallbad ( 4.7) gestellt und anschließend 15 min auf dem Schüttelgerät geschüttelt oder auf dem Magnetrührer gerührt ( 4.1). Die Lösung wird über Glasfaser-Filterpapier ( 4.2) filtriert.
Ein aliquoter Teil des Filtrats wird mit dem Wasser-Methanol-Acetonitril-Gemisch ( 3.12) verdünnt, bis eine endgültige Carbadoxkonzentration von 10 bis 15g/ml erreicht ist. (Verdünnungsfaktor 10 für eine 10 %ige Zubereitung). Ein aliquoter Teil wird durch einen 0,45-m-Membranfilter ( 4.6) filtriert.
Anschließend wird die HPLC-Bestimmung ( 5.4) durchgeführt.
5.3 Reinigung
5.3.1 Vorbereitung der Aluminiumoxid-Säule
Von Aluminiumoxid ( 3.4) werden 4 g abgewogen und in die Glassäule ( 4.3) eingebracht.
5.3.2 Reinigung der Probe
Von dem filtrierten Extrakt (5.2.1) werden 15 ml auf die Aluminiumoxid-Säule gegeben, und die ersten 2 ml des Eluats werden verworfen. Die folgenden 5 ml werden aufgefangen und ein aliquoter Teil davon durch einen 0,45-m-Membranfilter ( 4.6) filtriert.
Anschließend wird die HPLC-Bestimmung ( 5.4) durchgeführt.
5.4 HPLC-Bestimmung
5.4.1 Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren
| Ergebnissen führen: | |
| HPLC-Trennsäule ( 4.4.1): | 300 × 4 mm, C18, 10 µm Korngröße, oder vergleichbare Säule |
| Mobile Phase (3.10): | Gemisch aus Acetat-Pufferlösung (3.9) und Acetonitril ( 3.2), 825 + 175 (V +V) |
| Durchflussrate: | 1,5 bis 2 ml/min |
| Detektionswellenlänge: | 365 nm |
| Einspritzvolumen: | 20 µl |
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung (3.11.2), die 5,0 µg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erhalten werden.
5.4.2 Erstellung der Kalibrationskurve
Jede Kalibrierlösung ( 3.11.2) wird mehrmals eingespritzt und die Peakhöhen (-flächen) werden für die einzelnen Konzentrationen gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen (-flächen) auf der Ordinate und die dazugehörigen Konzentrationen in g/ml auf der Abszisse aufgetragen werden.
5.4.3 Bestimmung der Probenlösung
Der Probenextrakt [( 5.3.2) für Futtermittel, ( 5.2.2) für Vormischungen und ( 5.2.3) für Zubereitungen] wird mehrmals eingespritzt und die mittlere Peakhöhe (-fläche) der Carbadoxpeaks ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Carbadoxpeaks der Probenlösung wird anhand der Kalibrationskurve ( 5.4.2) die Carbadoxkonzentration der Probenlösung in µg/ml bestimmt.
6.1 Futtermittel
Der Carbadoxgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet: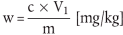
wobei:
c = Carbadoxkonzentration der Probenlösung ( 5.3.2) µg/ml,
V1 = Extraktionsvolumen in ml (d. h. 50),
m = Probeneinwaage in g.
6.2 Vormischungen und Zubereitungen
Der Carbadoxgehalt w (mg/kg) der Probe wird nach folgender Formel berechnet: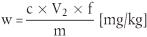
wobei:
c = Carbadoxkonzentration der Probenlösung ( 5.2.2 oder 5.2.3) in µg/ml,
V2 = Extraktionsvolumen in ml (d. h. 50 für Vormischungen; 150 für Zubereitungen),
f = Verdünnungsfaktor gemäß 5.2.2 (Vormischungen) oder 5.2.3(Zubereitungen),
m = Probeneinwaage in g.
7. Überprüfung der Ergebnisse
7.1 Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren des Probenextrakts und der Kalibrierlösung ( 3.11.2), die 10,0 µg/ml enthält, verglichen werden.
7.1.1 Co-Chromatografie
Ein Probenextrakt wird mit einer geeigneten Menge Kalibrierlösung ( 3.11.2) angereichert. Die zugesetzte Carbadoxmenge muss in etwa dem vermuteten Carbadoxgehalt des Probenextrakts entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Carbadoxpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks der nicht angereicherten Probenlösung abweichen.
7.1.2 Diodenarray-Detektion
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2 Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf bei einem Carbadoxgehalt von 10 mg/kg und mehr 15 % des höheren Wertes nicht überschreiten.
7.3 Wiederfindungsrate
Bei einer angereicherten (Blind-)Probe muss die Wiederfindungsrate mindestens 90 % betragen.
8. Ergebnisse eines Ringversuchs
Bei einem Ringversuch wurden 6 Futtermittel, 4 Vormischungen und 3 Zubereitungen von 8 Laboratorien untersucht. Jede Probe wurde doppelt analysiert. (Nähere Informationen zu diesem Ringversuch sind dem Journal of the AOAC, Band 71, 1988, S. 484-490, zu entnehmen.) Die Ergebnisse (mit Ausnahme von Ausreißern) sind in der nachstehenden Tabelle zusammengefasst:
Tabelle 1 : Ergebnisse des Ringversuchs für Futtermittel
| Probe 1 | Probe 2 | Probe 3 | Probe 4 | Probe 5 | Probe 6 | |
| L | 8 | 8 | 8 | 8 | 8 | 8 |
| n | 15 | 14 | 15 | 15 | 15 | 15 |
| Mittelwert [mg/kg] | 50,0 | 47,6 | 48,2 | 49,7 | 46,9 | 49,7 |
| sr[mg/kg] | 2,90 | 2,69 | 1,38 | 1,55 | 1,52 | 2,12 |
| VKr[ %] | 5,8 | 5,6 | 2,9 | 3,1 | 3,2 | 4,3 |
| sR[mg/kg] | 3,92 | 4,13 | 2,23 | 2,58 | 2,26 | 2,44 |
| VKR[ %] | 7,8 | 8,7 | 4,6 | 5,2 | 4,8 | 4,9 |
| Sollgehalt [mg/kg] | 50,0 | 50,0 | 50,0 | 50,0 | 50,0 | 50,0 |
Tabelle 2 : Ergebnisse des Ringversuchs für Vormischungen und Zubereitungen
| Vormischungen | Zubereitungen | ||||||
| A | B | C | D | A | B | C | |
| L | 7 | 7 | 7 | 7 | 8 | 8 | 8 |
| n | 14 | 14 | 14 | 14 | 16 | 16 | 16 |
| Mittelwert [mg/kg] | 8,89 | 9,29 | 9,21 | 8,76 | 94,6 | 98,1 | 104 |
| sr [mg/kg] | 0,37 | 0,28 | 0,28 | 0,44 | 4,1 | 5,1 | 7,7 |
| VKr[ %] | 4,2 | 3,0 | 3,0 | 5,0 | 4,3 | 5,2 | 7,4 |
| sR [mg/kg] | 0,37 | 0,28 | 0,40 | 0,55 | 5,4 | 6,4 | 7,7 |
| VKR[ %] | 4,2 | 3,0 | 4,3 | 6,3 | 5,7 | 6,5 | 7,4 |
| Sollgehalt [mg/kg] | 10,0 | 10,0 | 10,0 | 10,0 | 100 | 100 | 100 |
L = Anzahl der Laboratorien
n = Anzahl der Einzelwerte
sr = Standardabweichung der Wiederholbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
| Entsprechungstabellen gemäss Artikel 6 | Anhang IX |
1. Richtlinie 71/250 /EWG
| Richtlinie 71/250/EWG | Vorliegende Verordnung |
| Artikel 1 Unterabsatz 1 | Artikel 3 |
| Artikel 1 Unterabsatz 2 | Artikel 2 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang Teil 1 | Anhang II |
| Anhang Teil 2 | - |
| Anhang Teil 3 | - |
| Anhang Teil 4 | Anhang III Teil O |
| Anhang Teil 5 | Anhang III Teil M |
| Anhang Teil 6 | Anhang III Teil N |
| Anhang Teil 7 | Anhang III Teil Q |
| Anhang Teil 9 | Anhang III Teil K |
| Anhang Teil 10 | - |
| Anhang Teil 11 | - |
| Anhang Teil 12 | Anhang III Teil J |
| Anhang Teil 14 | Anhang III Teil D |
| Anhang Teil 16 | - |
2. Richtlinie 71/393 /EWG
| Richtlinie 71/393/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang Teil I | Anhang III Teil A |
| Anhang Teil II | Anhang III Teil E |
| Anhang Teil III | Anhang III Teil P |
| Anhang Teil IV | Anhang III Teil H |
3. Richtlinie 72/199 /EWG
| Richtlinie 72/199/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Anhang I Teil 1 | Anhang III Teil L |
| Anhang I Teil 2 | Anhang III Teil C |
| Anhang I Teil 2 | - |
| Anhang I Teil 3 | - |
| Anhang I Teil 3 | Anhang V Teil A |
| Anhang II | - |
4. Richtlinie 73/46 /EWG
| Richtlinie 73/46/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 3 | - |
| Artikel 4 | - |
| Anhang I Teil 1 | Anhang III Teil B |
| Anhang I Teil 2 | - |
| Anhang I Teil 3 | Anhang III Teil I |
5. Richtlinie 76/371/EWG
| Richtlinie 76/371/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 1 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | Anhang I |
6. Richtlinie 76/372/EWG
| Richtlinie 76/372/EWG | Vorliegende Verordnung |
| Artikel 1 | - |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | - |
7. Richtlinie 78/633 /EWG
| Richtlinie 78/633/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang Teil 1 | - |
| Anhang Teil 2 | - |
| Anhang Teil 3 | Anhang IV Teil C |
8. Richtlinie 81/715/EWG
| Richtlinie 81/715/EWG | Vorliegende Verordnung |
| Artikel 1 | - |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | - |
9. Richtlinie 84/425/EWG
| Richtlinie 84/425/EWG | Vorliegende Verordnung |
| Artikel 1 | - |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | - |
10. Richtlinie 86/174/EWG
| Richtlinie 86/174/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 4 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | Anhang VII |
11. Richtlinie 93/70/EWG
| Richtlinie 93/70/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | Anhang IV Teil D |
12. Richtlinie 93/117/EG
| Richtlinie 93/117/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 und 5 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang Teil 1 | Anhang IV Teil E |
| Anhang Teil 2 | Anhang VIII Teil A |
13. Richtlinie 98/64/EG
| Richtlinie 98/64/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 und 5 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Anhang Teil A | Anhang III Teil F |
| Anhang Teil C | Anhang VIII Teil B |
14. Richtlinie 1999/27/EG
| Richtlinie 1999/27/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 und 5 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Artikel 5 | - |
| Artikel 6 | - |
| Artikel 7 | - |
| Anhang Teil A | Anhang VIII Teil C |
| Anhang Teil B | Anhang IV Teil F |
| Anhang Teil C | Anhang VIII Teil D |
15. Richtlinie 1999/76/EG
| Richtlinie 1999/76/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Anhang | Anhang IV Teil G |
16. Richtlinie 2000/45/EG
| Richtlinie 2000/45/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Anhang Teil A | Anhang IV Teil A |
| Anhang Teil B | Anhang IV Teil B |
| Anhang Teil C | Anhang III Teil G |
17. Richtlinie 2002/70/EG
| Richtlinie 2002/70/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 1 |
| Artikel 2 | Artikel 2 und 3 |
| Artikel 3 | - |
| Artikel 4 | - |
| Artikel 5 | - |
| Anhang I | Anhang I und Anhang V Teil B(I) |
| Anhang II | Anhang II und Anhang V Teil B(II) |
18. Richtlinie 2003/126 /EG
| Richtlinie 2003/126/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Artikel 5 | - |
| Artikel 6 | - |
| Anhang | Anhang VI |
|
ENDE | |
(Stand: 29.10.2020)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: 90.- € netto (Grundlizenz)
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion