

umwelt-online: Verordnung (EG) Nr. 152/2009 zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln (4)
|
zurück | |
C. Bestimmung des Gehalts an den Spurenelementen Eisen, Kupfer, Mangan und Zink
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an den Spurenelementen Eisen, Kupfer, Mangan und Zink in Futtermitteln. Die Bestimmungsgrenzen liegen bei:
2. Prinzip
Die Probe wird nach Zerstörung eventuell vorhandener organischer Substanz in Salzsäure gelöst. Die Elemente Eisen, Kupfer, Mangan und Zink werden nach entsprechender Verdünnung der Analysenlösung mittels Atomabsorptionsspektrometrie bestimmt.
3. Reagenzien
Vorbemerkungen
Das zur Herstellung der Reagenzien und Analysenlösungen verwendete Wasser muss "frei" von den zu bestimmenden Kationen sein, d. h., das Wasser muss entweder in einer Borsilikatglas- oder Quarzapparatur 2-fach destilliert oder durch doppelten Ionenaustausch gereinigt worden sein.
Die zur Analyse verwendeten Reagenzien müssen mindestens von analysenreiner Qualität sein. Die Abwesenheit des zu bestimmenden Elements wird mittels eines Blindversuchs kontrolliert. Falls erforderlich, sind die Reagenzien einer zusätzlichen Reinigung zu unterziehen.
Anstelle der nachfolgend beschriebenen Standardlösungen können auch handelsübliche Standardlösungen verwendet werden, deren Reinheit garantiert und vor der Verwendung kontrolliert wurde.
3.1 Salzsäure (D: 1,19 g/ml).
3.2 Salzsäure (6 mol/l).
3.3 Salzsäure (0,5 mol/l).
3.4 Fluorwasserstoffsäure (Volumenkonzentration = 38 bis 40 %); Eisengehalt: weniger als 1 mg/l; Glührückstand (als Sulfat): weniger als 10 mg/l.
3.5 Schwefelsäure (D: 1,84 g/ml).
3.6 Wasserstoffperoxid, ca. 100 Volumina Sauerstoff (Massenanteil = 30 %).
3.7 Eisen-Standardlösung (1.000 µg Fe/ml), wie folgt zubereitet, oder eine gleichwertige handelsübliche Lösung: 1 g Eisendraht wird in 200 ml 6 mol/l Salzsäure (3.2) gelöst. Es werden 16 ml Wasserstoffperoxid ( 3.6) zugegeben; dann wird mit Wasser auf 1 l aufgefüllt:
3.7.1 Eisen-Gebrauchsstandardlösung (100 µg Fe/ml): Die Standardlösung ( 3.7) wird im Verhältnis 1:9 mit Wasser verdünnt.
3.8 Kupfer-Standardlösung (1.000 µg Cu/ml), wie folgt zubereitet, oder eine gleichwertige handelsübliche Lösung:
3.8.1 Kupfer-Gebrauchsstandardlösung (10 µg Cu/ml): Die Standardlösung (3.8) wird im Verhältnis 1:9 mit Wasser verdünnt; anschließend wird die so entstandene Lösung wiederum im Verhältnis 1:9 mit Wasser verdünnt.
3.9 Mangan-Standardlösung (1.000 µg Mn/ml), wie folgt zubereitet, oder eine gleichwertige handelsübliche Lösung:
3.9.1 Mangan-Gebrauchsstandardlösung (10 µg Mn/ml): Die Standardlösung (3.9) wird im Verhältnis 1:9 mit Wasser verdünnt; anschließend wird die so entstandene Lösung wiederum im Verhältnis 1:9 mit Wasser verdünnt.
3.10 Zink-Standardlösung (1.000 µg Zn/ml), wie folgt zubereitet, oder eine gleichwertige handelsübliche Lösung:
3.10.1 Zink-Gebrauchsstandardlösung (10 µg Zn/ml): Die Standardlösung ( 3.10) wird im Verhältnis 1:9 mit Wasser verdünnt; anschließend wird die so entstandene Lösung wiederum im Verhältnis 1:9 mit Wasser verdünnt.
3.11 Lanthanchloridlösung: In 150 ml Wasser wird 12 g Lanthanoxid gelöst und 100 ml 6 mol/l Salzsäure ( 3.2) zugegeben. Dann wird mit Wasser auf 1 l aufgefüllt.
4. Geräte
4.1 Muffelofen mit Regelvorrichtung und gegebenenfalls mit Temperaturanzeige.
4.2 Glasgeräte müssen aus resistentem Borsilikatglas sein. Es wird empfohlen, Glasgeräte zu benutzen, die ausschließlich der Bestimmung des Gehalts an Spurenelementen vorbehalten bleiben.
4.3 Atomabsorptions-Spektralfotometer; das Gerät muss innerhalb des vorgesehenen Messbereichs hinsichtlich seiner Empfindlichkeit und Genauigkeit den Anforderungen der jeweiligen Methode genügen.
5. Verfahren1
5.1 Proben mit organischen Bestandteilen
5.1.1 Veraschung und Herstellung der Analysenlösung2
5.1.1.1 Von der Probe werden 5 bis 10 g (auf 0,2 mg genau abgewogen) in eine Quarz- oder Platinschale gegeben (siehe Anmerkung b) und im Trockenschrank bei 105 °C getrocknet; dann wird die Schale in den kalten Muffelofen ( 4.1) verbracht. Der Ofen wird geschlossen (siehe Anmerkung c) und die Temperatur innerhalb von ca. 90 min langsam auf 450 bis 475 °C erhöht. Diese Temperatur wird 4 bis 16 h (z.B. über Nacht) aufrechterhalten, um Kohleteilchen zu entfernen; dann wird der Ofen geöffnet und abkühlen gelassen (siehe Anmerkung d).
Die Asche wird mit Wasser angefeuchtet und in ein 250-ml-Becherglas gegeben. Die Schale wird mit insgesamt etwa 5 ml Salzsäure ( 3.1) ausgespült, die anschließend langsam und vorsichtig (da es aufgrund von CO2-Bildung eventuell zu einer heftigen Reaktion kommen kann) in das Becherglas gegeben wird. Unter Schütteln wird Salzsäure ( 3.1) tropfenweise zugegeben, bis die Schaumbildung aufhört. Die Salzsäure wird unter gelegentlichem Umrühren mit einem Glasstab bis zur Trockne abgedampft.
Dem Rückstand werden 15 ml 6 mol/l Salzsäure ( 3.2) und anschließend etwa 120 ml Wasser zugefügt. Umgerührt wird mit dem Glasstab, der in dem Becherglas zu belassen ist. Letzteres wird mit einem Uhrglas abgedeckt. Die Flüssigkeit wird langsam zum Sieden gebracht und so lange im Sieden gehalten, bis die Asche vollständig gelöst ist. Dann wird durch ein aschefreies Filterpapier filtriert und das Filtrat in einem 250-ml-Messkolben aufgefangen. Das Becherglas und der Filter werden mit 5 ml heißer 6 mol/l Salzsäure ( 3.2) und 2-mal mit kochendem Wasser ausgewaschen. Anschließend wird der Messkolben mit Wasser zur Marke aufgefüllt (HCl-Konzentration etwa 0,5 mol/l).
5.1.1.2 Sollte der Filterrückstand schwarz aussehen (Kohlenstoff), so wird er im Trockenschrank abermals bei 450 bis 475 °C verascht. Diese Veraschung, die nur wenige Stunden erfordert (etwa 3 bis 5 h) ist abgeschlossen, wenn die Asche weiß oder nahezu weiß aussieht. Der Rückstand wird mit etwa 2 ml Salzsäure ( 3.1) aufgenommen, zur Trockne eingedampft und mit 5 ml 6 mol/l Salzsäure ( 3.2) versetzt. Nach dem Erwärmen wird die Lösung in den Messkolben filtriert, und Letzterer mit Wasser zur Marke aufgefüllt (HCl-Konzentration etwa 0,5 mol/l).
Anmerkungen:a. Bei der Bestimmung des Gehalts an Spurenelementen ist unbedingt auf die Gefahr von Verunreinigungen, insbesondere durch Zink, Kupfer und Eisen zu achten. Deshalb müssen die bei der Probenvorbereitung benutzten Geräte frei von diesen Metallen sein.
Um die Gefahr von Verunreinigungen einzuschränken, ist in staubfreier Luft, mit absolut reinen Geräten und sorgfältig gewaschenen Glasgeräten zu arbeiten. Die Zinkbestimmung ist für Verunreinigungen durch Glasgeräte, Reagenzien, Staub usw. besonders anfällig.
b. Die Einwaage wird nach dem etwa zu erwartenden Spurenelementgehalt des Futtermittels unter Berücksichtigung der Empfindlichkeit des verwendeten Spektralfotometers berechnet. Bei Futtermitteln, die einen niedrigen Gehalt an Spurenelementen aufweisen, kann es erforderlich sein, eine Probe von 10 bis 20 g einzuwägen und das Volumen der Analysenlösung auf 100 ml zu begrenzen.
c. Die Veraschung muss in einem geschlossenen Muffelofen ohne Einblasen von Luft oder Sauerstoff erfolgen.
d. Die vom Pyrometer angezeigte Temperatur darf 475 °C nicht überschreiten.
5.1.2 Spektralfotometrische Bestimmung
5.1.2.1 Herstellen der Kalibrierlösungen
Aus den Gebrauchsstandardlösungen 3.7.1, 3.8.1, 3.9.1 und 3.10.1 werden für jedes der zu bestimmenden Spurenelemente eine Reihe von Kalibrierlösungen hergestellt. Jede dieser Kalibrierlösungen hat eine HCl-Konzentration von etwa 0,5 mol/l und (im Falle von Eisen, Mangan und Zink) einen Lanthanchlorid-Gehalt, der einer Massenkonzentration von 0,1 % La entspricht.
Die gewählten Spurenelementkonzentrationen müssen im Empfindlichkeitsbereich des verwendeten Spektralfotometers liegen. Die nachfolgenden Tabellen zeigen Beispiele für die Zusammensetzung typischer Reihen von Kalibrierlösungen. Je nach Typ und Empfindlichkeit des verwendeten Spektralfotometers kann es jedoch erforderlich sein, für die Kalibrierlösungen eine andere Konzentration zu wählen.
Eisen
| µg Fe/ml | 0 | 0,5 | 1 | 2 | 3 | 4 | 5 |
| ml Gebrauchsstandardlösung ( 3.7.1) (1 ml = 100 µg Fe) |
0 | 0,5 | 1 | 2 | 3 | 4 | 5 |
| ml HCl ( 3.2) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| 10 ml Lanthanchloridlösung ( 3.11) zugeben und mit Wasser auf 100 ml auffüllen. | |||||||
Kupfer
| µg Cu/ml | 0 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| ml Gebrauchsstandardlösung ( 3.8.1) (1 ml = 10 µg Cu) |
0 | 1 | 2 | 4 | 6 | 8 | 10 |
| ml HCl ( 3.2) | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
Mangan
| µg Mn/ml | 0 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| ml Gebrauchsstandardlösung ( 3.9.1) (1 ml = 10 µg Mn) |
0 | 1 | 2 | 4 | 6 | 8 | 10 |
| ml HCl ( 3.2) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| 10 ml Lanthanchloridlösung ( 3.11) zugeben und mit Wasser auf 100 ml auffüllen. | |||||||
Zink
| µg Zn/ml | 0 | 0,05 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 |
| ml Gebrauchsstandardlösung ( 3.10.1) (1 ml = 10 µg Zn) |
0 | 0,5 | 1 | 2 | 4 | 6 | 8 |
| ml HCl ( 3.2) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| 10 ml Lanthanchloridlösung ( 3.11) zugeben und mit Wasser auf 100 ml auffüllen. | |||||||
5.1.2.2 Herstellen der Analysenlösung
Für die Bestimmung des Kupfergehalts kann die nach 5.1.1 hergestellte Analysenlösung in der Regel direkt verwendet werden. Gegebenenfalls wird ein aliquoter Teil dieser Analysenlösung in einen 100-ml-Messkolben pipettiert und mit 0,5 mol/l Salzsäure ( 3.3) zur Marke aufgefüllt, um ihre Konzentration in den Bereich der Kalibrierlösungen zu bringen.
Für die Bestimmung des Gehalts an Eisen, Mangan und Zink wird ein aliquoter Teil der nach 5.1.1 hergestellten Lösung in einen 100-ml-Messkolben pipettiert. Es werden 10 ml Lanthanchloridlösung ( 3.11) zugegeben und mit 0,5 mol/l Salzsäure ( 3.3) zur Marke aufgefüllt (siehe Bemerkung 8).
5.1.2.3 Blindversuch
Es wird ein Blindversuch ausgeführt, der alle Verfahrensschritte umfassen muss, nur dass das Probematerial selbst weggelassen wird. Die Kalibrierlösung "00 ersetzt nicht den Blindversuch.
5.1.2.4 Messung der Atomabsorption
Die Atomabsorption der Kalibrierlösungen und der Analysenlösungen ist mit oxydierender Luft-Azetylen-Flamme bei folgenden Wellenlängen zu messen:
Fe: 248,3 nm,
Cu: 324,8 nm,
Mn: 279,5 nm,
Zn: 213,8 nm.
Jede Messung ist 4-mal auszuführen.
5.2 Mineralfutter
Enthält die Probe keine organischen Substanzen, so erübrigt sich eine vorherige Veraschung. In diesem Fall wird dann ab 5.1.1.1 zweiter Absatz weiterverfahren. Ein Abrauchen mit Fluorwasserstoffsäure kann entfallen.
6. Berechnung der Ergebnisse
Die Spurenelementkonzentration in der Analysenlösung wird mithilfe einer Kalibrationskurve berechnet und das Ergebnis in mg Spurenelement/kg der Probe (ppm) ausgedrückt.
7. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen eines Analytikers bei ein und derselben Probe darf die folgenden Werte nicht überschreiten:
8. Bemerkung
Das Vorhandensein von größeren Phosphatmengen kann die Bestimmung des Gehalts an Eisen, Mangan und Zink beeinträchtigen. Diese Interferenzen sind durch die Zugabe von Lanthanchloridlösung ( 3.11) zu korrigieren. Weist die Probe jedoch das Gewichtsverhältnis Ca + Mg/P > 2 auf, kann auf die Zugabe von Lanthanchloridlösung ( 3.11) zu der Analysenlösung und den Kalibrierlösungen verzichtet werden.
________
1) Andere Aufschlussmethoden können verwendet werden, sofern erwiesen ist, dass sie zu vergleichbaren Ergebnissen führen (z.B.Mikrowellen-Druckaufschluss).
2) Grünfutter (frisch oder getrocknet) kann größere Mengen pflanzlicher Kieselsäure enthalten, welche Spurenelemente binden kann und daher entfernt werden muss. Bei Proben dieser Futtermittel muss also nach folgendem geänderten Verfahren vorgegangen werden: Das Verfahren 5.1.1.1 wird bis zur Filtration durchgeführt. Das den unlöslichen Rückstand enthaltende Filterpapier wird 2-mal mit kochendem Wasser ausgewaschen, in eine Quarz- oder Platinschale gegeben und im Muffelofen ( 4.1) bei weniger als 550 °C bis zur vollständigen Elimination aller Kohlepartikel verascht. Nach dem Abkühlen werden einige Tropfen Wasser und danach 10-15 ml Fluorwasserstoffsäure ( 3.4) zugegeben und bei ca. 150 °C zur Trockne eingedampft. Verbleibt im Rückstand noch Kieselsäure, so wird diese erneut in einigen ml Fluorwasserstoffsäure ( 3.4) gelöst und anschließend zur Trockne eingedampft. Dann werden 5 Tropfen Schwefelsäure ( 3.5) zugesetzt und so lange erhitzt, bis keine weißen Dämpfe mehr auftreten. Nach Zugabe von 5 ml 6 mol/l Salzsäure ( 3.2) und etwa 30 ml Wasser wird erhitzt und die Lösung in den 250-ml-Messkolben filtriert. Letzterer wird mit Wasser zur Marke aufgefüllt (HCl-Konzentration etwa 0,5 mol/l). Anschließend wird wie ab 5.1.2 beschrieben weiterverfahren.
D. Bestimmung des Halofuginongehalts
DL-trans-7-Brom-6-chlor-3-(3-(3-hydroxy-2-piperidyl)acetonyl)-4(3H)-chinazolinonhydrobromid
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Halofuginongehalts von Futtermitteln. Die Bestimmungsgrenze beträgt 1 mg/kg.
2. Prinzip
Nach der Behandlung mit heißem Wasser wird Halofuginon als freie base mit Ethylacetat extrahiert und anschließend durch Ausschütteln mit Salzsäure in das Hydrochlorid überführt. Der Extrakt wird durch Ionenaustauschchromatografie gereinigt. Der Halofuginongehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (HPLC) unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
3.1 Acetonitril, HPLC-Qualität.
3.2 Amberlite XAD-2-Harz.
3.3 Ammoniumacetat.
3.4 Ethylacetat.
3.5 Essigsäure (Eisessig).
3.6 Halofuginon-Standardsubstanz: DL-trans-7-Brom-6-chlor-3-[3-(3-hydroxy-2-piperidyl)acetonyl]-4(3H)-chinazolinon-hydrobromid, E 764
3.6.1 Halofuginon-Standard-Stammlösung, 100 µg/ml
Von Halofuginon ( 3.6) werden 50 mg auf 0,1 mg genau in einen 500-ml-Messkolben eingewogen und in Ammoniumacetat-Pufferlösung ( 3.18) gelöst. Es wird mit der Pufferlösung zur Marke aufgefüllt und gemischt. Diese Lösung ist 3 Wochen haltbar, wenn sie im Dunkeln bei 5 °C aufbewahrt wird.
3.6.2 Kalibrierlösungen
Von der Standard-Stammlösung ( 3.6.1) werden 1,0, 2,0, 3,0, 4,0 bzw. 6,0 ml jeweils in einen 100-ml-Messkolben überführt. Es wird mit der mobilen Phase ( 3.21) zur Marke aufgefüllt und durchmischt. Diese Lösungen enthalten Halofuginon in Konzentrationen von 1,0, 2,0, 3,0, 4,0 bzw. 6,0g/ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden;
3.7 Salzsäure (ρ20 = ca. 1,16 g/ml).
3.8 Methanol.
3.9 Silbernitrat.
3.10 Natriumascorbat.
3.11 Natriumcarbonat (Soda).
3.12 Natriumchlorid.
3.13 EDTa (Ethylendiamintetraessigsäure, Dinatriumsalz).
3.14 Wasser, HPLC-Qualität.
3.15 Natriumcarbonatlösung, c = 10 g/100 ml.
3.16 Mit Natriumchlorid gesättigte Natriumcarbonatlösung, c = 5 g/100 ml
50 g Natriumcarbonat ( 3.11) werden in Wasser gelöst und auf 1 l verdünnt; dann wird Natriumchlorid ( 3.12) zugegeben, bis die Lösung gesättigt ist.
3.17 Salzsäure, ca. 0,1 mol/l
10 ml Salzsäure ( 3.7) werden mit Wasser auf 1 l verdünnt.
3.18 Ammoniumacetat-Pufferlösung, ca. 0,25 mol/l
19,3 g Ammoniumacetat ( 3.3) und 30 ml Essigsäure ( 3.5) werden in Wasser ( 3.14) gelöst und auf 1 l verdünnt.
3.19. Vorbereitung des Amberlite XAD-2-Harzes
Eine ausreichende Menge Amberlite ( 3.2) wird mit Wasser chloridfrei gewaschen. Die Prüfung der Waschflüssigkeit erfolgt mit Silbernitratlösung (3.20). Danach wird das Harz mit 50 ml Methanol ( 3.8) gewaschen. Das Methanol wird verworfen und das Harz in frischem Methanol aufbewahrt.
3.20 Silbernitratlösung, ca. 0,1 mol/l
0,17 g Silbernitrat ( 3.9) werden in 10 ml Wasser gelöst.
3.21 Mobile Phase für die HPLC:
500 ml Acetonitril (3.1) werden mit 300 ml Ammoniumacetat-Pufferlösung ( 3.18) und 1.200 ml Wasser ( 3.14) gemischt. Der pH-Wert wird mit Essigsäure ( 3.5) auf 4,3 eingestellt. Die Lösung wird durch einen 0,22-µm-Filter ( 4.8) filtriert und entgast (z.B. durch 10-minütige Ultraschallbehandlung). Diese Lösung ist 1 Monat lang haltbar, wenn sie in einem verschlossenen Gefäß im Dunkeln aufbewahrt wird.
4. Geräte
4.1 Ultraschallbad.
4.2 Rotationsverdampfer.
4.3 Zentrifuge.
4.4 HPLC-Einrichtung mit UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor
4.4.1 HPLC-Trennsäule, 300 × 4 mm, C18, 10 µm Korngröße, oder vergleichbare Säule.
4.5 Glassäule (300 × 10 mm) mit gesintertem Glasfilter und Absperrhahn.
4.6 Glasfaserfilter, 150 mm Durchmesser.
4.7 Membranfilter, 0,45 µm Porengröße.
4.8 Membranfilter, 0,22 µm Porengröße.
5. Verfahren
Anmerkung:
Halofuginon ist als freie base in Alkali- und Ethylacetat-Lösungen instabil. Es darf höchstens 30 min in Ethylacetat bleiben.
5.1 Allgemeines
5.1.1 Zur Prüfung, dass weder Halofuginon noch Störsubstanzen vorhanden sind, ist eine Blindprobe zu untersuchen.
5.1.2 Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Halofuginon angereichert wurde. Die zugesetzte Menge an Halofuginon sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 3 mg/kg werden 300 µl der Standard-Stammlösung ( 3.6.1) zu 10 g der Blindprobe gegeben. Es wird gemischt und 10 min gewartet, bevor mit der Extraktion ( 5.2) fortgefahren wird.
Anmerkung:
Für den Zweck dieser Methode muss die Blindprobe ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Halofuginon darf nicht nachweisbar sein.
5.2 Extraktion
Von der vorbereiteten Probe werden 10 g auf 0,1 g genau in ein 200-ml-Zentrifugenglas eingewogen. Es werden 0,5 g Natriumascorbat ( 3.10), 0,5 g EDTa ( 3.13) und 20 ml Wasser hinzugefügt, und es wird gemischt. Das Zentrifugenglas wird 5 min in ein 80 °C heißes Wasserbad gestellt. Nach dem Abkühlen auf Raumtemperatur werden 20 ml Natriumcarbonatlösung ( 3.15) hinzugefügt, und es wird gemischt. Unmittelbar danach werden 100 ml Ethylacetat ( 3.4) hinzugefügt, und es wird 15 s kräftig von Hand geschüttelt. Danach wird das Zentrifugenglas mit gelockertem Stopfen 3 min in ein Ultraschallbad ( 4.1) gestellt. Es wird 2 min zentrifugiert und die Ethylacetatphase durch einen Glasfaserfilter ( 4.6) in einen 500-ml-Scheidetrichter dekantiert. Die Extraktion der Probe wird mit weiteren 100 ml Ethylacetat wiederholt. Die vereinigten Extrakte werden 1 min lang mit 50 ml der mit Natriumchlorid gesättigten Natriumcarbonatlösung ( 3.16) gewaschen. Die wässrige Phase wird verworfen.
Die organische Phase wird 1 min mit 50 ml Salzsäure ( 3.17) extrahiert. Die untere Säurephase wird in einen 250-ml-Scheidetrichter abgelassen. Die organische Phase wird erneut 1,5 min mit weiteren 50 ml Salzsäure extrahiert. Die beiden Säureextrakte werden vereinigt und durch ca. 10 s langes Schütteln mit 10 ml Ethylacetat ( 3.4) gewaschen.
Die wässrige Phase wird quantitativ in einen 250-ml-Rundkolben überführt und die organische Phase verworfen. Das restliche in der sauren Lösung enthaltene Ethylacetat wird mithilfe des Rotationsverdampfers ( 4.2) entfernt. Die Temperatur des Wasserbads darf 40 °C nicht überschreiten. Bei einem Unterdruck von ca. 25 mbar wird das restliche Ethylacetat bei 38 °C innerhalb von 5 min entfernt.
5.3 Cleanup
5.3.1 Vorbereitung der Amberlitesäule
Für jeden Probenextrakt wird eine XAD-2-Säule vorbereitet. Von dem vorbereiteten Amberlite ( 3.19) werden 10 g mit Methanol ( 3.8) in eine Glassäule ( 4.5) eingefüllt. Auf das obere Ende des Harzbettes wird ein kleiner Glaswattebausch gebracht. Das Methanol wird aus der Säule ablaufen gelassen und das Harz mit 100 ml Wasser gewaschen. Sobald die Flüssigkeit das obere Ende des Harzbettes erreicht hat, wird der Absperrhahn geschlossen. Vor Gebrauch ist die Säule 10 min zu äquilibrieren. Die Säule darf nie trockenlaufen.
5.3.2 Cleanup der Probe
Der Extrakt ( 5.2) wird quantitativ auf die vorbereitete Amberlitesäule ( 5.3.1) aufgebracht und eluiert. Das Eluat wird verworfen. Die Elutionsgeschwindigkeit darf 20 ml/min nicht überschreiten. Der Rundkolben wird mit 20 ml Salzsäure ( 3.17) gespült und die Austauschersäule mit der Spülflüssigkeit gewaschen. Die eventuell auf der Säule verbliebene saure Lösung wird mit einem Luftstrom restlos ausgeblasen. Die Waschlösungen werden verworfen. Es werden 100 ml Methanol ( 3.8) auf die Säule gegeben und ein Eluat von 5 bis 10 ml in einem 250-ml-Rundkolben aufgefangen. Das restliche Methanol wird 10 min lang zur Äquilibrierung auf dem Harz belassen; anschließend wird die Elution mit einer Elutionsgeschwindigkeit von höchstens 20 ml/min fortgesetzt und das Eluat in demselben Rundkolben aufgefangen. Das Methanol wird am Rotationsverdampfer ( 4.2) abgedampft, wobei die Temperatur des Wasserbads 40 °C nicht übersteigen darf. Der Rückstand wird unter Verwendung der mobilen Phase ( 3.21) quantitativ in einen 10-ml-Messkolben überführt. Es wird mit der mobilen Phase zur Marke aufgefüllt und gemischt. Ein aliquoter Teil wird durch einen Membranfilter ( 4.7) filtriert. Diese Lösung wird für die HPLC-Bestimmung ( 5.4) aufbewahrt.
5.4 HPLC-Bestimmung
5.4.1 Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
HPLC-Trennsäule ( 4.4.1),
Mobile Phase für die HPLC ( 3.21),
Durchflussrate: 1,5 bis 2 ml/min,
Detektionswellenlänge: 243 nm,
Einspritzvolumen: 40 bis 100 µl.
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung ( 3.6.2), die 3,0 µg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.4.2 Erstellung der Kalibrationskurve
Jede Kalibrierlösung ( 3.6.2) wird mehrmals eingespritzt, und die Peakhöhen (-flächen) für die einzelnen Konzentrationen werden gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen oder -flächen der Kalibrierlösungen auf der Ordinate und die dazugehörigen Konzentrationen ing/ml auf der Abszisse aufgetragen werden.
5.4.3 Bestimmung der Probenlösung
Der Probenextrakt ( 5.3.2) wird mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird, und die mittlere Peakhöhe (-fläche) der Halofuginonpeaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Peakhöhe (-fläche) der Halofuginonpeaks der Probenlösung wird anhand der Kalibrationskurve ( 5.4.2) die Konzentration der Probenlösung in µg/ml bestimmt.
Der Halofuginongehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
| w = | c x 10 |
| m |
wobei:
c = Halofuginonkonzentration der Probenlösung in µg/ml,
m = Probeneinwaage in g.
7. Überprüfung der Ergebnisse
7.1 Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung und der Kalibrierlösung ( 3.6.2), die 6,0 µg/ml enthält, verglichen werden.
7.1.1 Co-Chromatografie
Eine Probenlösung wird mit einer geeigneten Menge einer Kalibrierlösung ( 3.6.2) versetzt. Die Menge des zugesetzten Halofuginons muss dem erwarteten Halofuginongehalt der Probenlösung entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Halofuginonpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks der nicht angereicherten Probenlösung abweichen.
7.1.2 Diodenarray-Detektion
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2 Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf bei einem Halofuginongehalt von bis zu 3 mg/kg 0,5 mg/kg nicht überschreiten.
7.3 Wiederfindungsrate
Für eine angereicherte Blindprobe muss die Wiederfindung mindestens 80 % betragen.
8. Ergebnisse eines Ringversuchs
Es wurde ein Ringversuch1 durchgeführt, bei dem 3 Proben von 8 Laboratorien untersucht wurden. Die Ergebnisse sind in der nachstehenden Tabelle zusammengefasst:
Ergebnisse
| Probe A (Blindprobe) nach Erhalt |
Probe B (Mehl) | Probe C (Pellets) | |||
| Nach Erhalt | Nach 2 Monaten | Nach Erhalt | Nach 2 Monaten | ||
| Mittelwert [mg/kg] | NN | 2,80 | 2,42 | 2,89 | 2,45 |
| sR[mg/kg] | - | 0,45 | 0,43 | 0,40 | 0,42 |
| VKR[ %] | - | 16 | 18 | 14 | 17 |
| Wiederfindung [ %] | 86 | 74 | 88 | 75 | |
NN = nicht nachweisbar
sR = Standardabweichung der Vergleichbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
_____
1) The Analyst 108, 1983, S. 1252-1256.
E. Bestimmung des Robenidingehalts
1,3-bis[(4-Chlorobenzyliden)amino]-guanidin-hydrochlorid
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Robenidingehalts von Futtermitteln. Die Bestimmungsgrenze beträgt 5 mg/kg.
2. Prinzip
Die Probe wird mit angesäuertem Methanol extrahiert. Der Extrakt wird getrocknet und ein aliquoter Teil zum Cleanup auf eine Aluminiumoxidsäule gegeben. Robenidin wird mit Methanol von der Säule eluiert, eingeengt und mit der mobilen Phase auf ein geeignetes Volumen gebracht. Der Robenidingehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (HPLC) unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
3.1 Methanol
3.2 Angesäuertes Methanol
In einen 500-ml-Messkolben werden 4,0 ml Salzsäure (ρ20 = 1,18 g/ml) überführt. Es wird mit Methanol ( 3.1) zur Marke aufgefüllt und durchmischt. Diese Lösung muss vor Gebrauch frisch hergestellt werden.
3.3 Acetonitril, HPLC-Qualität
3.4 Molekularsieb
Typ 3A, 8 bis 12 Mesh (Korngröße 1,6 bis 2,5 mm, kristallines Aluminiumsilicat, Porendurchmesser 0,3 nm).
3.5 Aluminiumoxid, sauer, Aktivitätsstufe I, für die Säulenchromatografie
100 g Aluminiumoxid werden in ein geeignetes Gefäß gegeben und mit 2,0 ml Wasser versetzt. Das Gefäß wird verschlossen und etwa 20 min geschüttelt. Das Aluminiumoxid ist in einem gut verschlossenen Behälter aufzubewahren.
3.6 Kaliumdihydrogenphosphatlösung, c = 0,025 mol/l
In einem 1.000-ml-Messkolben werden 3,40 g Kaliumdihydrogenphosphat in Wasser (HPLC-Qualität) gelöst. Es wird zur Marke aufgefüllt und durchmischt.
3.7 Dinatriumhydrogenphosphatlösung, c = 0,025 mol/l
In einem 1 l-Messkolben werden 3,55 g wasserfreies Dinatriumhydrogenphosphat (oder 4,45 g Dihydrat oder 8,95 g Dodecahydrat) in Wasser (HPLC-Qualität) gelöst. Es wird zur Marke aufgefüllt und durchmischt.
3.8 Mobile Phase für die HPLC
Folgende Reagenzien werden gemischt:
650 ml Acetonitril ( 3.3),
250 ml Wasser (HPLC-Qualität),
50 ml Kaliumdihydrogenphosphatlösung ( 3.6),
50 ml Dinatriumhydrogenphosphatlösung ( 3.7).
Die Lösung wird durch einen 0,22-µm-Filter ( 4.6) filtriert und entgast (z.B. durch 10-minütige Ultraschallbehandlung).
3.9 Standardsubstanz
Robenidin 1,3-bis[(4-Chlorobenzyliden)amino]-guanidinhydrochlorid, rein
3.9.1 Robenidin-Standard-Stammlösung, 300 µg/ml
Von der Robenidin-Standardsubstanz ( 3.9) werden 30 mg auf 0,1 mg genau eingewogen, in einem 100-ml-Messkolben in angesäuertem Methanol ( 3.2) gelöst und mit demselben Lösungsmittel zur Marke aufgefüllt und durchmischt. Der Messkolben wird mit Aluminiumfolie umhüllt und im Dunkeln aufbewahrt.
3.9.2 Robenidin-Standardlösung, 12 µg/ml
Von der Standard-Stammlösung ( 3.9.1) werden 10,0 ml in einen 250-ml-Messkolben überführt. Es wird mit der mobilen Phase (3.8) zur Marke aufgefüllt und durchmischt. Der Messkolben wird mit Aluminiumfolie umhüllt und im Dunkeln aufbewahrt.
3.9.3 Kalibrierlösungen
Von der Robenidin-Standardlösung ( 3.9.2) werden 5,0, 10,0, 15,0, 20,0 bzw. 25,0 ml jeweils in einen 50-ml-Messkolben überführt. Es wird mit der mobilen Phase ( 3.8) zur Marke aufgefüllt und durchmischt. Diese Lösungen enthalten Robenidin in Konzentrationen von 1,2, 2,4, 3,6, 4,8 bzw. 6,0 µg/ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden.
3.10 Wasser, HPLC-Qualität
4. Geräte
4.1 Glassäule
Braunglas mit Absperrhahn und Reservoir mit einem Fassungsvermögen von ca. 150 ml, Innendurchmesser 10 bis 15 mm, Länge 250 mm.
4.2 Mechanischer Schüttler oder Magnetrührer.
4.3 Rotationsfilmverdampfer.
4.4 HPLC-Einrichtung mit UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor mit einem Messbereich von 250 bis 400 nm
4.4.1 HPLC-Trennsäule: 300 × 4 mm, C18, 10 µm Korngröße, oder vergleichbare Säule.
4.5 Glasfaser-Filterpapier, z.B. Whatman GF/a oder gleichwertig.
4.6 Membranfilter, 0,22 µm Porengröße.
4.7 Membranfilter, 0,45 µm Porengröße.
5. Verfahren
Anmerkung:
Robenidin ist lichtempfindlich. Bei allen Verfahrensschritten sind Braunglasgeräte zu verwenden.
5.1 Allgemeines
5.1.1 Zur Prüfung, dass weder Robenidin noch Störsubstanzen vorhanden sind, ist eine Blindprobe zu untersuchen.
5.1.2 Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe ( 5.1.1) untersucht wird, die mit Robenidin angereichert wurde. Die zugesetzte Menge an Robenidin sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 60 mg/kg werden 3,0 ml der Standard-Stammlösung ( 3.9.1) in einen 250-ml-Erlenmeyerkolben gegeben. Die Lösung wird in einem Stickstoffstrom auf ca. 0,5 ml eingeengt. Dann werden 15 g der Blindprobe zugegeben. Es wird gemischt und 10 min stehen gelassen, bevor mit der Extraktion ( 5.2) fortgefahren wird.
Anmerkung:
Für den Zweck dieser Methode muss die Blindprobe ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Robenidin darf nicht nachweisbar sein.
5.2 Extraktion
Von der vorbereiteten Probe werden 15 g auf 0,01 g genau in einen 250-ml-Erlenmeyerkolben eingewogen, mit 100,0 ml angesäuertem Methanol ( 3.2) versetzt und nach Verschließen des Kolbens 1 h auf dem Schüttler ( 4.2) geschüttelt. Die Lösung wird über Glasfaserfilterpapier ( 4.5) filtriert und das gesamte Filtrat in einem 150-ml-Erlenmeyerkolben aufgefangen. Es werden 7,5 g Molekularsieb ( 3.4) zugegeben, und nach Verschließen des Kolbens wird 5 min geschüttelt. Anschließend wird sofort durch ein Glasfaserfilterpapier filtriert. Diese Lösung wird für die Reinigung ( 5.3) aufbewahrt.
5.3 Reinigung
5.3.1 Vorbereitung der Aluminiumoxid-Säule
In das untere Ende der Glassäule ( 4.1) wird ein Glaswattebausch eingebracht, der mit einem Glasstab zusammengedrückt wird. Von dem vorbereiteten Aluminiumoxid ( 3.5) werden 11,0 g auf die Säule aufgebracht. Dabei ist darauf zu achten, dass der Kontakt mit der Luft so gering wie möglich gehalten wird. Durch leichtes Klopfen an das untere Ende der befüllten Säule wird das Aluminiumoxid verdichtet.
5.3.2 Reinigung der Probe
Von dem Probenextrakt ( 5.2) werden 5,0 ml mit einer Pipette auf die Säule gegeben, wobei die Pipettenspitze die Säulenwand berühren soll. Nach Absorption der Lösung durch das Aluminiumoxid wird das Robenidin mit 100 ml Methanol ( 3.1) bei einer Flussrate von 2-3 ml/min von der Säule eluiert und das Eluat in einem 250-ml-Rundkolben aufgefangen. Die Methanollösung wird am Rotationsverdampfer ( 4.3) bei 40 °C und unter vermindertem Druck bis zur Trockne eingeengt. Der Rückstand wird mit 3 bis 4 ml der mobilen Phase ( 3.8) erneut gelöst und quantitativ in einen 10-ml-Messkolben überführt. Der Kolben wird mehrmals mit je 1 bis 2 ml der mobilen Phase gespült, und die Spüllösungen werden in den Messkolben gegeben. Es wird mit der mobilen Phase zur Marke aufgefüllt und gemischt. Ein aliquoter Teil wird durch einen 0,45-m-Membranfilter ( 4.7) filtriert. Diese Lösung wird für die HPLC-Bestimmung ( 5.4) aufbewahrt.
5.4 HPLC-Bestimmung
5.4.1 Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
HPLC-Trennsäule ( 4.4.1),
mobile Phase für die HPLC ( 3.8),
Durchflussrate: 1,5 bis 2 ml/min,
Detektionswellenlänge: 317 nm,
Einspritzvolumen: 20 bis 50 µl).
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung ( 3.9.3), die 3,6 µg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.4.2 Erstellung der Kalibrationskurve
Jede Kalibrierlösung ( 3.9.3) wird mehrmals eingespritzt, und die Peakhöhen (-flächen) für die einzelnen Konzentrationen werden gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen (-flächen) der Kalibrierlösungen auf der Ordinate und die dazugehörigen Konzentrationen ing/ml auf der Abszisse aufgetragen werden.
5.4.3 Bestimmung der Probenlösung
Der Probenextrakt ( 5.3.2) wird mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird, und die mittlere Peakhöhe (-fläche) der Robenidinpeaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Robenidinpeaks der Probenlösung wird anhand der Kalibrationskurve ( 5.4.2) die Konzentration der Probenlösung ing/ml bestimmt.
Der Robenidingehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
| w = | c x 200 |
| m |
wobei:
c = Robenidinkonzentration der Probenlösung in µg/ml,
m = Probeneinwaage in g.
7. Überprüfung der Ergebnisse
7.1 Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung und der Kalibrierlösung ( 3.9.3), die 6 µg/ml enthält, verglichen werden.
7.1.1 Co-Chromatografie
Eine Probenlösung wird mit einer geeigneten Menge Kalibrierlösung ( 3.9.3) versetzt. Die Menge des zugesetzten Robenidins muss dem erwarteten Robenidingehalt der Probenlösung entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Robenidinpeaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des Peaks der nicht angereicherten Probenlösung abweichen.
7.1.2 Diodenarray-Detektion
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2. Wiederholbarkeit
Bei einem Robenidingehalt von mehr als 15 mg/kg darf die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe 10 % des höheren Werts nicht überschreiten.
7.3 Wiederfindungsrate
Für eine angereicherte Blindprobe muss die Wiederfindungsrate mindestens 85 % betragen.
8. Ergebnisse eines Ringversuchs
Bei einem EG-Ringversuch wurden 4 Proben von Geflügel- und Kaninchenfutter in Form von Mehl oder Pellets von 12 Laboratorien analysiert. Jede Probe wurde doppelt analysiert. Die Ergebnisse sind in der nachstehenden Tabelle zusammengefasst:
| Geflügelfutter | Kaninchenfutter | |||
| Mehl | Pellets | Mehl | Pellets | |
| Mittelwert [mg/kg] | 27,00 | 27,99 | 43,6 | 40,1 |
| sr[mg/kg] | 1,46 | 1,26 | 1,44 | 1,66 |
| VKr[ %] | 5,4 | 4,5 | 3,3 | 4,1 |
| sR[mg/kg] | 4,36 | 3,36 | 4,61 | 3,91 |
| VKR[ %] | 16,1 | 12,0 | 10,6 | 9,7 |
| Wiederfindung [ %] | 90,0 | 93,3 | 87,2 | 80,2 |
sr = Standardabweichung der Wiederholbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
F. Bestimmung des Diclazurilgehalts
2,6-Dichlor-alpha-(4-chlorophenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3H)-yl)benzacetonitril
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Diclazurilgehalts von Futtermitteln und Vormischungen. Die Nachweisgrenze beträgt 0,1 mg/kg, die Bestimmungsgrenze 0,5 mg/kg.
2. Prinzip
Nach Hinzufügen eines internen Standards wird die Probe mit angesäuertem Methanol extrahiert. Bei Futtermitteln wird ein aliquoter Teil des Extrakts mit einer C18-Festphasen-Extraktionskartusche gereinigt. Diclazuril wird aus der Kartusche mit einem Gemisch aus angesäuertem Methanol und Wasser eluiert. Nach dem Einengen wird der Rückstand in DMF/Wasser gelöst. Bei Vormischungen wird der Extrakt eingeengt und der Rückstand in DMF/Wasser gelöst. Der Diclazurilgehalt wird mittels ternärer Gradienten-Hochleistungsflüssigkeitschromatografie (HPLC) über eine Umkehrphase unter Verwendung eines UV-Detektors bestimmt.
3. Reagenzien
3.1 Wasser, HPLC-Qualität.
3.2 Ammoniumacetat.
3.3 Tetrabutylammoniumhydrogensulfat (TBHS).
3.4 Acetonitril, HPLC-Qualität.
3.5 Methanol, HPLC-Qualität.
3.6 N,N-Dimethylformamid (DMF).
3.7 Salzsäure, ρ20 = 1,19 g/ml.
3.8 Standardsubstanz: Diclazuril II-24: 2,6-Dichlor-alpha-(4-chlorophenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3H)-yl)benzacetonitril, rein, E 771.
3.8.1 Diclazuril-Standard-Stammlösung, 500 µg/ml
Von der Diclazuril-Standardsubstanz ( 3.8) werden 25 mg auf 0,1 mg genau in einen 50-ml-Messkolben eingewogen und in DMF ( 3.6) gelöst; es wird mit DMF ( 3.6) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 °C ist diese Lösung 1 Monat haltbar.
3.8.2 Diclazuril-Standardlösung, 50 µg/ml
Von der Standard-Stammlösung ( 3.8.1) werden 5,00 ml in einen 50-ml-Messkolben überführt; es wird mit DMF ( 3.6) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 °C ist diese Lösung 1 Monat haltbar.
3.9 Interne Standardsubstanz: 2,6 Dichloro-α-(4-chlorophenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3H)-yl)-α-methylbenzacetonitril
3.9.1 Interne Standard-Stammlösung, 500 µg/ml
Von der internen Standardsubstanz ( 3.9) werden 25 mg auf 0,1 mg genau in einen 50-ml-Messkolben eingewogen und in DMF ( 3.6) gelöst; es wird mit DMF (3.6) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤4 °C ist diese Lösung 1 Monat haltbar.
3.9.2 Interne Standardlösung, 50 µg/ml
Von der internen Standard-Stammlösung ( 3.9.1.) werden 5,00 ml in einen 50-ml-Messkolben überführt; es wird mit DMF ( 3.6) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 °C ist diese Lösung 1 Monat haltbar.
3.9.3 Interne Standardlösung für Vormischungen, p/1.000 mg/ml
(p = Diclazuril-Sollgehalt in der Vormischung in mg/kg)
Von der internen Standardsubstanz werden p/10 mg auf 0,1 mg genau in einen 100-ml-Messkolben eingewogen und mittels Ultraschallbad ( 4.6) in DMF ( 3.6) gelöst; es wird mit DMF ( 3.6) zur Marke aufgefüllt und gemischt. Der Messkolben wird mit Aluminiumfolie umhüllt (oder ein Messkolben aus braunem Glas verwendet) und im Kühlschrank aufbewahrt. Bei einer Temperatur von ≤ 4 °C ist diese Lösung 1 Monat haltbar.
3.10 Kalibrierlösung, 2 µg/ml
In einen 50-ml-Messkolben werden 2,00 ml Diclazuril-Standardlösung ( 3.8.2) und 2,00 ml interne Standardlösung ( 3.9.2) pipettiert. Es werden 16 ml DMF ( 3.6) hinzugefügt; anschließend wird mit Wasser zur Marke aufgefüllt und gemischt. Diese Lösung muss vor Gebrauch frisch hergestellt werden.
3.11 C18-Festphasen-Extraktionskartusche, z.B. Bond Elut, Größe: 1 cc, Sorptionsmenge: 100 mg.
3.12 Extraktionslösung: angesäuertes Methanol
5,0 ml Salzsäure ( 3.7) werden in 1.000 ml Methanol (3.5) pipettiert und gemischt.
3.13 Mobile Phase für die HPLC:
3.13.1 Elutionsmittel A: Ammoniumacetat-Tetrabutylammoniumhydrogensulfat-Lösung,
5 g Ammoniumacetat ( 3.2) und 3,4 g TBHS ( 3.3) werden in 1.000 ml Wasser (3.1) gelöst und gemischt,
3.13.2 Elutionsmittel B: Acetonitril ( 3.4),
3.13.3 Elutionsmittel C: Methanol ( 3.5).
4. Geräte
4.1 Mechanischer Schüttler.
4.2 HPLC-Einrichtung für ternären Gradienten
4.2.1 HPLC-Trennsäule, 3m Korngröße, 100 × 4,6 mm, z.B. Hypersil ODS, oder vergleichbare Säule.
4.2.2 UV-Detektor mit variabler Wellenlängeneinstellung oder Diodenarray-Detektor.
4.3 Rotationsfilmverdampfer.
4.4 Membranfilter, 0,45 µm Porengröße.
4.5 Vakuumeinrichtung.
4.6 Ultraschallbad.
5. Verfahren
5.1 Allgemeines
5.1.1 Blindprobe
Zur Prüfung, dass weder Diclazuril noch Störsubstanzen vorhanden sind, ist eine Blindprobe zu untersuchen. Die Blindprobe muss ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Diclazuril oder Störsubstanzen dürfen nicht nachweisbar sein.
5.1.2 Wiederfindungstest
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Diclazuril angereichert wurde. Die zugesetzte Menge sollte der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 1 mg/kg werden 0,1 ml der Standard-Stammlösung ( 3.8.1) zu 50 g der Blindprobe gegeben. Es wird gründlich gemischt, 10 min stehen gelassen und nochmals mehrfach gemischt, bevor mit der Extraktion (5.2) fortgefahren wird.
Ist eine der zu untersuchenden Probe ähnliche Blindprobe nicht verfügbar (siehe 5.1.1), so kann ein Wiederfindungstest mithilfe des Additionsverfahrens durchgeführt werden. In diesem Fall wird die zu untersuchende Probe mit einer Diclazurilmenge angereichert, die der bereits in der Probe vorhandenen Menge entspricht. Diese Probe wird zusammen mit der nicht angereicherten Probe untersucht, und die Wiederfindungsrate kann durch Subtraktion ermittelt werden.
5.2 Extraktion
5.2.1 Futtermittel
Von der Probe werden 50 g auf 0,01 g genau eingewogen und in einen 500-ml-Erlenmeyerkolben überführt. Es werden 1,00 ml der internen Standardlösung ( 3.9.2) und 200 ml Extraktionslösung ( 3.12) hinzugefügt. Anschließend wird der Kolben verschlossen. Die Mischung wird über Nacht auf dem Schüttler ( 4.1) geschüttelt und anschließend 10 min stehen gelassen. Ein aliquoter Teil von 20 ml der überstehenden Lösung wird in einen geeigneten Glasbehälter überführt und mit 20 ml Wasser verdünnt. Diese Lösung wird auf eine Extraktionskartusche ( 3.11) gegeben und mittels Vakuum ( 4.5) hindurch gesaugt. Die Kartusche wird mit 25 ml einer Mischung aus Extraktionslösung ( 3.12) und Wasser, 65 + 35 (V+V), ausgewaschen. Die gesammelten Fraktionen werden verworfen, und der Wirkstoff und der interne Standard werden mit 25 ml einer Mischung aus Extraktionslösung ( 3.12) und Wasser, 80 + 20 (V+V), eluiert. Diese Fraktion wird mithilfe eines Rotationsverdampfers ( 4.3) bei 60 °C zur Trockne eingeengt. Der Rückstand wird in 1,0 ml DMF ( 3.6) gelöst; es werden 1,5 ml Wasser ( 3.1) hinzugefügt, gemischt und durch einen Membranfilter (4.4) filtriert. Anschließend wird die HPLC-Bestimmung ( 5.3) durchgeführt.
5.2.2 Vormischungen
Von der Probe wird 1 g auf 0,001 g genau eingewogen und in einen 500-ml-Erlenmeyerkolben überführt. Es werden 1,00 ml interne Standardlösung ( 3.9.3) und 200 ml Extraktionslösung ( 3.12) hinzugefügt, und der Kolben wird verschlossen. Die Mischung wird über Nacht auf dem Schüttler ( 4.1) geschüttelt und anschließend 10 min stehen gelassen. Ein 10.000/pml-Aliquot (p = Diclazuril-Sollgehalt in der Vormischung in mg/kg) der überstehenden Lösung wird in einen Rundkolben geeigneter Größe überführt. Es wird mithilfe eines Rotationsverdampfers (4.3) bei vermindertem Druck und 60 °C zur Trockne eingeengt. Der Rückstand wird in 10,0 ml DMF ( 3.6) gelöst; es werden 15,0 ml Wasser ( 3.1) hinzugefügt und gemischt. Anschließend wird die HPLC-Bestimmung ( 5.3) durchgeführt.
5.3 HPLC-Bestimmung
5.3.1 Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
| HPLC-Trennsäule ( 4.2.1.): |
100 × 4,6 mm, Hypersil ODS, 3 µm Korngröße, oder vergleichbare Säule |
|
| Mobile Phase: | Elutionsmittel a ( 3.13.1): | wässrige Lösung von Ammoniumacetat und Tetrabutylammonium-Hydrogensulfat |
| Elutionsmittel B ( 3.13.2): | Acetonitril | |
| Elutionsmittel C ( 3.13.3): | Methanol | |
| Elutionsmodus: | - linearer Gradient | |
| - Anfangsbedingungen: a + B + C = 60 + 20 + 20 (V+V+V) | ||
| - nach 10 min Gradientenelution 30 min lang: a + B + C = 45 + 20 + 35 (V+V+V) | ||
| Mit Elutionsmittel B 10 min spülen. | ||
| Durchflussrate: | 1,5 bis 2 ml/min | |
| Einspritzvolumen: | 20 µl | |
| Detektionswellenlänge: | 280 nm | |
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung ( 3.10), die 2 µg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.3.2 Bestimmung der Kalibrierlösung
Von der Kalibrierlösung ( 3.10) werden 20 µl mehrmals eingespritzt, und für Diclazuril und den internen Standard wird die mittlere Peakhöhe (-fläche) bestimmt.
5.3.3 Bestimmung der Probenlösung
Von der Probenlösung ( 5.2.1 oder 5.2.2) werden mehrmals 20 µl eingespritzt, und für Diclazuril und den internen Standard wird die mittlere Peakhöhe (-fläche) bestimmt.
6. Berechnung der Ergebnisse
6.1 Futtermittel
Der Diclazurilgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet:
wobei:
hd,s = Peakhöhe (-fläche) von Diclazuril in der Probenlösung ( 5.2.1),
hi,s = Peakhöhe (-fläche) des internen Standards in der Probenlösung ( 5.2.1),
hd,c = Peakhöhe (-fläche) von Diclazuril in der Kalibrierlösung ( 3.10),
hi,c = Peakhöhe (-fläche) des internen Standards in der Kalibrierlösung ( 3.10),
cd,c = Diclazurilkonzentration in der Kalibrierlösung in µg/ml ( 3.10),
m = Probeneinwaage in g,
V = Volumen der Probenextrakts gemäß 5.2.1 (d. h. 2,5 ml).
6.2 Vormischungen
Der Diclazurilgehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet: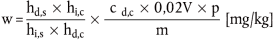
wobei:
hd,c = Peakhöhe (-fläche) von Diclazuril in der Kalibrierlösung ( 3.10),
hi,c = Peakhöhe (-fläche) des internen Standards in der Kalibrierlösung ( 3.10),
hd,s = Peakhöhe (-fläche) von Diclazuril in der Probenlösung (5.2.2),
hi,s = Peakhöhe (-fläche) des internen Standards in der Probenlösung ( 5.2.2),
cd,c = Diclazurilkonzentration in der Kalibrierlösung ing/ml ( 3.10),
m = Probeneinwaage in g,
V = Volumen des Probenextrakts gemäß 5.2.2 (d. h. 25 ml),
p = Diclazuril-Sollgehalt in der Vormischung in mg/kg.
7. Überprüfung der Ergebnisse
7.1 Identität
Die Identität des Analyten kann durch Co-Chromatografie oder mithilfe eines Diodenarray-Detektors bestätigt werden, wobei die Spektren der Probenlösung ( 5.2.1 oder 5.2.2) und der Kalibrierlösung ( 3.10) verglichen werden.
7.1.1 Co-Chromatografie
Eine nach 5.2.1 oder 5.2.2 hergestellte Probenlösung wird mit einer geeigneten Menge Kalibrierlösung ( 3.10) angereichert. Die Menge des zugesetzten Diclazurils muss dem erwarteten Diclazurilgehalt der Probenlösung entsprechen.
Unter Berücksichtigung der zugesetzten Menge und der Verdünnung des Extrakts darf nur die Höhe des Diclazurilpeaks und des Peaks des internen Standards vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der des ursprünglichen Diclazurilpeaks oder Standardpeaks des nicht angereicherten Probenextrakts abweichen.
7.1.2 Diodenarray-Detektion
Die Ergebnisse werden gemäß den nachstehenden Kriterien beurteilt:
Wird eines dieser Kriterien nicht erfüllt, gilt das Vorhandensein des Analyten als nicht bestätigt.
7.2 Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf folgende Werte nicht überschreiten:
Bei einer angereicherten (Blind-)Probe muss die Wiederfindungsrate mindestens 80 % betragen.
8. Ergebnisse eines Ringversuchs
Bei einem Ringversuch wurden 5 Proben von 11 Laboratorien untersucht. Diese Proben bestanden aus 2 Vormischungen, von denen eine mit einer organischen Matrix (O 100) und die andere mit einer anorganischen Matrix (a 100) gemischt war. Der theoretische Diclazurilgehalt liegt bei 100 mg/kg. Die 3 Geflügelmischfuttermittel stammten von 3 verschiedenen Herstellern (NL) (L1/Z1/K1). Der theoretische Diclazurilgehalt liegt bei 1 mg/kg. Die Laboratorien wurden beauftragt, jede der Proben 1- oder 2-mal zu untersuchen. (Nähere Informationen zu diesem Ringversuch sind dem Journal of AOAC International, Band 77, Nr. 6, 1994, S. 1359-1361, zu entnehmen.) Die Ergebnisse sind in der nachstehenden Tabelle zusammengefasst:
| Probe 1 a 100 |
Probe 2 O 100 |
Probe 3 L1 |
Probe 4 Z1 |
Probe 5 K1 |
|
| L | 11 | 11 | 11 | 11 | 6 |
| n | 19 | 18 | 19 | 19 | 12 |
| Mittelwert | 100,8 | 103,5 | 0,89 | 1,15 | 0,89 |
| sr (mg/kg) | 5,88 | 7,64 | 0,15 | 0,02 | 0,03 |
| VKr (%) | 5,83 | 7,38 | 17,32 | 1,92 | 3,34 |
| sR (mg/kg) | 7,59 | 7,64 | 0,17 | 0,11 | 0,12 |
| VKR (%) | 7,53 | 7,38 | 18,61 | 9,67 | 13,65 |
| Sollgehalt (mg/kg) | 100 | 100 | 1 | 1 | 1 |
L = Anzahl der Laboratorien
n = Anzahl der Einzelwerte
sr = Standardabweichung der Wiederholbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
9. Bemerkungen
Es muss vorab erwiesen sein, dass das Diclazurilsignal im linearen Konzentrationsbereich liegt.
G. Bestimmung des Gehalts an Lasalocid-Natrium
Monocarboxylsäure-Polyether-Natriumsalz, gebildet durch Streptomyces lasaliensis
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an Lasalocid-Natrium in Futtermitteln und Vormischungen. Die Nachweisgrenze beträgt 5 mg/kg, die Bestimmungsgrenze 10 mg/kg.
2. Prinzip
Lasalocid-Natrium wird aus der Probe mit angesäuertem Methanol extrahiert und mittels Umkehrphasen-Hochleistungsflüssigchromatografie (RP-HPLC) unter Verwendung eines spektrofluorometrischen Detektors bestimmt.
3. Reagenzien
3.1 Kaliumdihydrogenphosphat (KH2 PO4).
3.2 Orthophosphorsäure, w (Massenanteil) = 85 %.
3.3 Orthophosphorsäurelösung, c = 20 %
23,5 ml Orthophosphorsäure ( 3.2) werden mit Wasser auf 100 ml aufgefüllt.
3.4 6-Methyl-2-Heptylamin (1,5-Dimethylhexylamin), w (Massenanteil) = 99 %.
3.5 Methanol, HPLC-Qualität.
3.6 Salzsäure, Dichte = 1,19 g/ml.
3.7 Phosphatpufferlösung, c = 0,01 mol/l
In 500 ml Wasser ( 3.11) werden 1,36 g KH2PO4 ( 3.1) gelöst, und es werden 3,5 ml Orthophosphorsäure ( 3.2) sowie 10,0 ml 6-Methyl-2-Heptylamin ( 3.4) hinzugefügt. Den pH-Wert mit Orthophosphorsäurelösung ( 3.3) auf pH 4,0 einstellen und mit Wasser ( 3.11) auf 1.000 ml auffüllen.
3.8 Angesäuertes Methanol
In einen 1.000-ml-Messkolben werden 5,0 ml Salzsäure ( 3.6) gegeben, und es wird mit Methanol ( 3.5) zur Marke aufgefüllt und gemischt. Diese Lösung muss vor Gebrauch frisch hergestellt werden.
3.9 Mobile Phase für die HPLC: Phosphatpuffer-Methanollösung 5 + 95 (V+V)
Von der Phosphatpufferlösung ( 3.7) werden 5 ml mit 95 ml Methanol ( 3.5) vermischt.
3.10 Lasalocid-Natrium-Standardsubstanz, garantiert rein, C34H53O8Na (Monocarboxylsäure-Polyether-Natriumsalz, gebildet durch Streptomyces lasaliensis), E 763
3.10.1 Lasalocid-Natrium-Standard-Stammlösung, 500 µg/ml
Von Lasalocid-Natrium ( 3.10) werden 50 mg auf 0,1 mg genau in einen 100-ml-Messkolben eingewogen und in angesäuertem Methanol ( 3.8) gelöst. Es wird mit demselben Lösungsmittel zur Marke aufgefüllt und gemischt. Diese Lösung muss vor Gebrauch frisch hergestellt werden.
3.10.2 Lasalocid-Natrium-Standardlösung, 50 µg/ml
Von der Standard-Stammlösung ( 3.10.1) werden 10,0 ml in einen 100-ml-Messkolben pipettiert; es wird mit angesäuertem Methanol ( 3.8) zur Marke aufgefüllt und gemischt. Diese Lösung muss vor Gebrauch frisch hergestellt werden.
3.10.3 Kalibrierlösungen
Von der Lasalocid-Natrium-Standardlösung ( 3.10.2) werden 1,0, 2,0, 4,0, 5,0 bzw. 10,0 ml in jeweils einen 50-ml-Messkolben überführt. Es wird mit angesäuertem Methanol ( 3.8) zur Marke aufgefüllt und durchmischt. Diese Kalibrierlösungen enthalten jeweils 1,0, 2,0, 4,0, 5,0 bzw. 10,0 µg Lasalocid-Natrium je ml. Die Lösungen müssen vor Gebrauch frisch hergestellt werden.
3.11 Wasser, HPLC-Qualität.
4. Geräte
4.1 Ultraschallbad (oder Schüttel-Wasserbad) mit Temperatursteuerung.
4.2 Membranfilter, 0,45 µm Porengröße.
4.3 HPLC-Einrichtung mit Injektionssystem für Einspritzvolumina von 20 µl
4.3.1 HPLC-Trennsäule, 125 × 4 mm, mit Umkehrphase C18, 5 µm Korngröße, oder vergleichbare Säule.
4.3.2 Spektrofluorometer mit variabler Einstellung für Anregungs- und Emissionswellenlängen.
5. Verfahren
5.1 Allgemeines
5.1.1 Blindprobe
Für die Durchführung des Wiederfindungstests ( 5.1.2) ist zur Prüfung, dass weder Lasalocid-Natrium noch Störsubstanzen vorhanden sind, eine Blindprobe zu untersuchen. Die Blindprobe muss ähnlich zusammengesetzt sein wie die zu untersuchende Probe, und Lasalocid-Natrium oder Störsubstanzen dürfen nicht nachweisbar sein.
5.1.2 Wiederfindungstest
Die Wiederfindungsrate wird ermittelt, indem eine Blindprobe untersucht wird, die mit Lasalocid-Natrium angereichert wurde. Die zugesetzte Menge sollte in etwa der in der Probe vorhandenen Menge entsprechen. Zur Anreicherung auf einen Gehalt von 100 mg/kg werden 10,0 ml der Standard-Stammlösung ( 3.10.1) in einen 250-ml-Erlenmeyerkolben überführt. Die Lösung wird auf ca. 0,5 ml eingeengt. Dann werden 50 g der Blindprobe zugegeben. Es wird gründlich gemischt, 10 min stehen gelassen und erneut mehrmals gemischt, bevor mit der Extraktion ( 5.2) fortgefahren wird.
Ist eine der zu untersuchenden Probe ähnliche Blindprobe nicht verfügbar (siehe 5.1.1), so kann ein Wiederfindungstest mithilfe des Additionsverfahrens durchgeführt werden. In diesem Fall wird die zu untersuchende Probe mit einer Lasalocid-Natrium-Menge angereichert, die in etwa der bereits in der Probe vorhandenen Menge entspricht. Diese Probe wird zusammen mit der nicht angereicherten Probe untersucht und die Wiederfindungsrate kann durch Subtraktion ermittelt werden.
5.2 Extraktion
5.2.1 Futtermittel
Von der Probe werden 5 bis 10 g auf 0,01 g genau in einen 250-ml-Erlenmeyerkolben mit Stopfen eingewogen und 100,0 ml angesäuertes Methanol ( 3.8) mit einer Pipette hinzugefügt. Der Stopfen wird lose aufgesetzt und der Inhalt zum Dispergieren geschwenkt. Der Kolben wird 20 min in ein Ultraschallbad ( 4.1) mit einer Temperatur von ca. 40 °C gestellt, dann entnommen und auf Raumtemperatur abkühlen gelassen. Der Kolben wird etwa 1 h stehen gelassen, bis die Schwebstoffe sich abgesetzt haben. Dann wird ein aliquoter Teil über einen 0,45-µm-Membranfilter ( 4.2) in ein geeignetes Gefäß filtriert. Anschließend wird die HPLC-Bestimmung ( 5.3) durchgeführt.
5.2.2 Vormischungen
Rund 2 g der nicht gemahlenen Vormischung werden auf 0,001 g genau in einen 250-ml-Messkolben gegeben. Es werden 100,0 ml angesäuertes Methanol ( 3.8) hinzugefügt; der Inhalt wird zum Dispergieren geschwenkt. Der Kolben wird mit Inhalt 20 min lang in ein Ultraschallbad ( 4.1) mit einer Temperatur von ca. 40 °C gestellt, dann entnommen und auf Raumtemperatur abkühlen gelassen. Es wird mit angesäuertem Methanol ( 3.8) zur Marke aufgefüllt und sorgfältig gemischt. Der Kolben wird etwa 1 h stehen gelassen, bis die Schwebstoffe sich abgesetzt haben. Dann wird ein aliquoter Teil durch einen 0,45-µm-Membranfilter ( 4.2) filtriert. Ein aliquoter Teil des klaren Filtrats wird mit angesäuertem Methanol ( 3.8) verdünnt, um eine Lösung mit einer Konzentration von etwa 4 µg/ml Lasalocid-Natrium zu erhalten. Anschließend wird die HPLC-Bestimmung ( 5.3) durchgeführt.
5.3 HPLC-Bestimmung
5.3.1 Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
| HPLC-Trennsäule (4.3.1): |
125 × 4 mm, Umkehrphase C18, 5 µm Korngröße, oder vergleichbare Säule |
| Mobile Phase (3.9) Durchflussrate: |
Mischung aus Phosphatpufferlösung ( 3.7) und Methanol ( 3.5), 5 + 95 (V+V) 1,2 ml/min |
| Detektionswellen- länge: |
|
| Anregung: | 310 nm |
| Emission | 419 nm |
| Einspritzvolumen: | 20 µl |
Die Stabilität des chromatografischen Systems wird überprüft, indem die Kalibrierlösung ( 3.10.3), die 4,0 µg/ml enthält, mehrmals eingespritzt wird, bis konstante Peakhöhen (-flächen) und Retentionszeiten erreicht sind.
5.3.2 Erstellung der Kalibrationskurve
Jede Kalibrierlösung ( 3.10.3) wird mehrmals eingespritzt, und es werden die mittleren Peakhöhen (-flächen) für die einzelnen Konzentrationen gemessen. Es wird eine Kalibrationskurve erstellt, indem die mittleren Peakhöhen (-flächen) auf der Ordinate und die dazugehörigen Konzentrationen ing/ml auf der Abszisse aufgetragen werden.
5.3.3 Bestimmung der Probenlösung
Die nach 5.2.1 bzw. 5.2.2 gewonnenen Probenextrakte werden mehrmals eingespritzt, wobei dasselbe Volumen wie für die Einspritzung der Kalibrierlösungen verwendet wird. Die mittlere Peakhöhe (-fläche) der Lasalocid-Natrium-Peaks wird ermittelt.
6. Berechnung der Ergebnisse
Aus der mittleren Peakhöhe (-fläche) der Probenlösung ( 5.3.3) wird anhand der Kalibrationskurve die Konzentration an Lasalocid-Natrium (µg/ml) bestimmt.
6.1 Futtermittel
Der Gehalt an Lasalocid-Natrium w (in mg/kg) der Probe wird nach folgender Formel berechnet: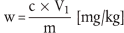
wobei:
c = Lasalocid-Natrium-Konzentration der Probenlösung ( 5.2.1) in µg/ml,
V1 = Volumen des Probenextrakts gemäß 5.2.1 (d. h. 100 ml),
m = Probeneinwaage in g.
6.2 Vormischungen
Der Gehalt an Lasalocid-Natrium w (in mg/kg) der Probe wird nach folgender Formel berechnet: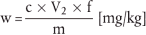
wobei:
c = Lasalocid-Natrium-Konzentration der Probenlösung ( 5.2.2) in µg/ml,
V2 = Volumen des Probenextrakts gemäß 5.2.2 in ml (d. h. 250 ml),
f = Verdünnungsfaktor gemäß 5.2.2,
m = Probeneinwaage in g.
7. Überprüfung der Ergebnisse
7.1 Identität
Nachweis- und Bestimmungsverfahren, denen die Fluoreszenzdetektion zugrunde liegt, sind im Vergleich zur UV-Detektion weniger interferenzanfällig. Die Identität des Analyten kann durch Co-Chromatografie bestätigt werden.
7.1.1 Co-Chromatografie
Ein Probenextrakt ( 5.2.1 oder 5.2.2) wird mit einer entsprechenden Menge Kalibrierlösung ( 3.10.3) angereichert. Die zugesetzte Lasalocid-Natrium-Menge muss in etwa dem Lasalocid-Natrium-Gehalt des Probenextrakts entsprechen. Unter Berücksichtigung der zugesetzten Lasalocid-Natrium-Menge und der Verdünnung des Extrakts darf nur die Höhe des Lasalocid-Natrium-Peaks vergrößert sein. Die Peakbreite in halber Höhe darf höchstens ± 10 % von der ursprünglichen Peakbreite abweichen, die sich bei dem nicht angereicherten Probenextrakt ergibt.
7.2 Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
7.3 Wiederfindungsrate
Bei einer angereicherten (Blind-)Probe muss die Wiederfindungsrate bei Futtermitteln mindestens 80 % betragen. Bei angereicherten Vormischungsproben muss die Wiederfindungsrate mindestens 90 % betragen.
8. Ergebnisse eines Ringversuchs
In einem Ringversuch * wurden 2 Vormischungen (Proben 1 und 2) und 5 Futtermittel (Proben 3 bis 7) in 12 Laboratorien untersucht. Jede Probe wurde doppelt analysiert. Die Ergebnisse sind in nachstehender Tabelle zusammengefasst:
| Probe 1 Vormi- schung Hühner- futter |
Probe 2 Vormi- schung Tuthahn- futter |
Probe 3 Trut- hahn- pellets |
Probe 4 Hühner- krümel- futter |
Probe 5 Trut- hahn- futter |
Probe 6 Geflügel- futter A |
Probe 7 Geflügel- futter B |
|
| L | 12 | 12 | 12 | 12 | 12 | 12 | 12 |
| n | 23 | 23 | 23 | 23 | 23 | 23 | 23 |
| Mittelwert [mg/kg] |
5 050 | 16 200 | 76,5 | 78,4 | 92,9 | 48,3 | 32,6 |
| sr[mg/kg] | 107 | 408 | 1,71 | 2,23 | 2,27 | 1,93 | 1,75 |
| VKr[ %] | 2,12 | 2,52 | 2,24 | 2,84 | 2,44 | 4,00 | 5,37 |
| sR[mg/kg] | 286 | 883 | 3,85 | 7,32 | 5,29 | 3,47 | 3,49 |
| VKR[ %] | 5,66 | 5,45 | 5,03 | 9,34 | 5,69 | 7,18 | 10,70 |
| Sollehalt [mg/kg] |
5 000 ) | 16 000 * | 80 * | 105 * | 120 * | 50 ** | 35 ** |
| *) Gehalt nach Angabe des Herstellers. **) Im Laboratorium zubereitetes Futter. |
|||||||
L = Anzahl der Laboratorien
n = Anzahl der Einzelwerte
sr = Standardabweichung der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
_____________
*) Analyst, 1995, 120, S. 2175-2180.
|
weiter . | |
(Stand: 29.10.2020)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: 90.- € netto (Grundlizenz)
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion