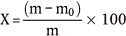
Verordnung (EG) Nr. 152/2009 der Kommission vom 27. Januar 2009 zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln
(Text von Bedeutung für den EWR)
(ABl. Nr. L 54 vom 26.02.2009 S. 1;
VO (EU) 278/2012 - ABl. Nr. L 91 vom 29.03.2012 S. 8Inkrafttreten Gültig;
VO (EU) 51/2013 - ABl. Nr. L 20 vom 23.01.2013 S. 33Inkrafttreten;
VO (EU) 691/2013 - ABl. Nr. L 197 vom 20.07.2013 S. 1Inkrafttreten Gültig;
VO (EU) 709/2014 - ABl. Nr. L 188 vom 27.06.2014 S. 1Inkrafttreten;
VO (EU) 2017/645 - ABl. Nr. L 115 vom 04.05.2017 S. 22 *;
VO (EU) 2017/771 - ABl. Nr. L 115 vom 04.05.2017 S. 22Inkrafttreten)
Neufassung -Ersetzt die RL'n *
Die Änderung betrifft nicht die deutsche Fassung.
| Hinweis: s. | Empf. (EU) 2016/1110 - (ABl. Nr. L 183 vom 08.07.2016 S. 68) |
| Empf. 2013/165/EU - (ABl. Nr. L 91 vom 03.04.2013 S. 12) |
RL'en 71/250/EWG, 71/393/EWG, 72/199/EWG, 73/46/EWG, 76/371/EWG, 76/372/EWG, 78/633/EWG, 81/715/EWG, 84/425/EWG, 86/174/EWG, 93/70/EWG, 93/117/EG, 98/64/EG, 1999/27/EG, 1999/76/EG, 2000/45/EG, 2002/70/EG und 2003/126/EG - Entsprechungstabelle - Anwenden
Die Kommission der Europäischen Gemeinschaften -
gestützt auf den Vertrag zur Gründung der Europäischen Gemeinschaft,
gestützt auf die Verordnung (EG) Nr. 882/2004 des Europäischen Parlaments und des Rates vom 29. April 2004 über amtliche Kontrollen zur Überprüfung der Einhaltung des Lebensmittel- und Futtermittelrechts sowie der Bestimmungen über Tiergesundheit und Tierschutz 1, insbesondere auf Artikel 11 Absatz 4 Buchstaben a, b und c,
in Erwägung nachstehender Gründe:
(1) Die nachstehenden Rechtsakte wurden zur Durchführung der Richtlinie 70/373/EWG erlassen und bleiben gemäß Artikel 61 Absatz 2 der Verordnung (EG) Nr. 882/2004 in Kraft:
(2) Da die Richtlinie 70/373/EWG durch die Verordnung (EG) Nr. 882/2004 ersetzt wurde, ist es angezeigt, die Durchführungsrechtsakte zu dieser Richtlinie aufzuheben und in einer einzigen Verordnung zusammenzufassen. Gleichzeitig sollten die Methoden und Verfahren entsprechend den neuesten wissenschaftlichen und technologischen Erkenntnissen angepasst werden. Methoden, die zu ihrem Zweck nicht mehr verwendet werden, sollten gestrichen werden. Es ist geplant, die Probenahmebestimmungen zu gegebener Zeit zu aktualisieren, um den jüngsten Entwicklungen bei der Erzeugung, Lagerung, Beförderung und Vermarktung von Futtermitteln Rechnung zu tragen. Dennoch ist es angezeigt, die bestehenden Probenahmebestimmungen vorläufig beizubehalten.
(3) Die Richtlinien 71/250/EWG, 71/393/EWG, 72/199/EWG, 73/46/EWG, 76/371/EWG, 76/372/EWG, 78/633/EWG, 81/715/EWG, 84/425/EWG, 86/174/EWG, 93/70/EWG, 93/117/EG, 98/64/EG, 1999/27/EG, 1999/76/EG, 2000/45/EG, 2002/70/EG und 2003/126/EG sollten daher aufgehoben werden.
(4) Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Ausschusses für die Lebensmittelkette und Tiergesundheit
- hat folgende Verordnung erlassen:
Die Probenahmen für die amtliche Untersuchung von Futtermitteln, insbesondere auf ihre Bestandteile, eingeschlossen Material, das genetisch veränderte Organismen (GVO) enthält, aus ihnen besteht oder aus ihnen hergestellt ist, Futtermittelzusatzstoffe gemäß der Verordnung (EG) Nr. 1831/2003 des Europäischen Parlaments und des Rates 20 und unerwünschte Stoffe gemäß der Richtlinie 2002/32/EG des Europäischen Parlaments und des Rates 21, erfolgen nach den in Anhang I aufgeführten Verfahren.
Das in Anhang I beschriebene Probenahmeverfahren gilt für die Untersuchung von Futtermitteln auf Pflanzenschutzmittelrückstände gemäß der Verordnung (EG) Nr. 396/2005 des Europäischen Parlaments und des Rates 22 und für die Überprüfung der Einhaltung der Verordnung (EU) Nr. 619/2011.
Die Vorbereitung der Analyseproben und die Formulierung der Ergebnisse erfolgen nach den in Anhang II aufgeführten Methoden.
Die Analyse für die amtliche Untersuchung von Futtermitteln erfolgt nach den Methoden, die in Anhang III(Analysemethoden zur Untersuchung der Zusammensetzung von Futtermittel-Ausgangserzeugnissen und Mischfuttermitteln), Anhang IV (Analysemethoden zur Untersuchung von Futtermitteln auf ihren Gehalt an zugelassenen Zusatzstoffen), Anhang V (Analysemethoden zur Untersuchung von Futtermitteln auf unerwünschte Stoffe) und Anhang VI (Analysemethoden zur Bestimmung der Bestandteile tierischen Ursprungs bei der amtlichen Untersuchung von Futtermitteln) aufgeführt sind.
Der Energiegehalt von Mischfuttermitteln für Geflügel wird in Übereinstimmung mit Anhang VII berechnet.
Die in Anhang VIII aufgeführten Analysemethoden für die Untersuchung auf rechtswidriges Vorhandensein von nicht mehr zugelassenen Zusatzstoffen in Futtermitteln werden zu Bestätigungszwecken verwendet.
Die Richtlinien 71/250/EWG, 71/393/EWG, 72/199/EWG, 73/46/EWG, 76/371/EWG, 76/372/EWG, 78/633/EWG, 81/715/EWG, 84/425/EWG, 86/174/EWG, 93/70/EWG, 93/117/EG, 98/64/EG, 1999/27/EG, 1999/76/EG, 2000/45/EG, 2002/70/EG und 2003/126/EG werden aufgehoben.
Verweise auf die aufgehobenen Richtlinien gelten als Verweise auf die vorliegende Verordnung nach den Entsprechungstabellen in Anhang IX.
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Sie gilt ab dem 26. August 2009.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 27. Januar 2009
| Probenahmeverfahren | Anhang I13 |
1. Zweck und Anwendungsbereich
Die zur amtlichen Untersuchung bestimmten Futtermittelproben werden gemäß den nachstehenden Verfahren entnommen. Die so gewonnenen Proben gelten als repräsentativ für die Teilpartien.
Zweck der repräsentativen Beprobung ist es, einen kleinen Teil einer Partie zu untersuchen und durch die Bestimmung eines spezifischen Merkmals bei diesem Teil den Durchschnittswert des Merkmals für die gesamte Partie zu ermitteln. Die Partie wird mittels wiederholter Entnahme von Einzelproben an verschiedenen Stellen der Partie untersucht. Diese Einzelproben werden zu einer Sammelprobe gemischt, aus der durch repräsentative Teilung repräsentative Endproben herzustellen sind.
Wenn bei der Sichtprüfung Teile des zu untersuchenden Futtermittels einen Qualitätsunterschied zum übrigen Futtermittel derselben Partie aufweisen, werden diese Teile vom übrigen Futtermittel abgesondert und als getrennte Teilpartie behandelt. Ist eine Aufteilung des Futtermittels in getrennte Teilpartien nicht möglich, so wird das Futtermittel als eine einzige Partie beprobt. In solchen Fällen ist dies im Probenahmebericht zu vermerken.
Ist ein Futtermittel, das gemäß den Bestimmungen der vorliegenden Verordnung beprobt wurde und nachweislich die EU-Anforderungen nicht erfüllt, Teil einer Futtermittelpartie derselben Gruppe oder Bezeichnung, so muss davon ausgegangen werden, dass das Ergebnis der Beprobung für die gesamte Futtermittelpartie gilt, es sei denn, eine eingehende Bewertung erbringt keinen Nachweis darüber, dass die restliche Partie den EU-Anforderungen nicht genügt.
2. Begriffsbestimmungen
3. Allgemeine Bestimmungen
4. Geräte
4.1 Die Geräte zur Probenahme müssen aus Materialien bestehen, die die zu beprobenden Erzeugnisse nicht kontaminieren können. Geräte, die für eine mehrfache Anwendung vorgesehen sind, müssen leicht zu reinigen sein, damit eine Kreuzkontamination vermieden wird.
4.2. Empfohlene Geräte für die Probenahme fester Futtermittel
4.2.1. Manuelle Probenahme
4.2.1.1 Schaufel mit ebenem Boden und rechteckig hochgebogenem Rand.
4.2.1.2 Probenahmestab mit langem Schlitz oder Kammern. Die Dimensionierung des Probenahmestabes ist den Merkmalen der Teilpartie (Tiefe des Behälters, Größe des Sacks usw.) und der Partikelgröße des Futtermittels anzupassen.
Falls es sich um einen Probenahmestab mit mehreren Öffnungen handelt, sollten diese durch Kammern oder fortlaufend versetzte Öffnungen voneinander getrennt sein, damit sichergestellt ist, dass die Probe an verschiedenen Stellen entlang des Probenahmestabes entnommen wird.
4.2.2. Mechanische Probenahme
Geeignete mechanische Geräte dürfen zur Probenahme aus in Bewegung befindlichen Futtermitteln verwendet werden. ,Geeignet' bedeutet, dass mindestens der gesamte Fließquerschnitt beprobt wird.
Die Probenahme aus in Bewegung befindlichen Futtermitteln (bei hohen Durchflussraten) kann mittels automatischer Probenehmer erfolgen.
4.2.3. Probenteiler
Soweit möglich und sinnvoll, sollten zur Herstellung reduzierter Proben Geräte verwendet werden, die zur Zerlegung der Probe in ungefähr gleiche Teile in repräsentativer Form bestimmt sind.
5. Quantitative Anforderungen hinsichtlich der Anzahl von Einzelproben
5.1. Quantitative Anforderungen an Einzelproben bezüglich der Untersuchung auf Stoffe oder Erzeugnisse, die gleichmäßig im Futtermittel verteilt sind
5.1.1. Feste Futtermittel in loser Form
| Teilpartie | Mindestanzahl der Einzelproben |
| ≤ 2,5 t | 7 |
| > 2,5 t | √20-mal die Anzahl von Tonnen, aus denen die Teilpartie besteht *, bis zu 40 Einzelproben |
| *) Wenn sich hierbei eine Bruchzahl ergibt, ist diese auf die nächsthöhere ganze Zahl aufzurunden. | |
5.1.2. Flüssige Futtermittel in loser Form
| Teilpartie | Mindestanzahl der Einzelproben |
≤ 2,5 t bzw. ≤ 2 500 l |
4 * |
| > 2,5 t bzw. > 2 500 l | 7 * |
| *) Ist eine Homogenisierung der Flüssigkeit nicht möglich, so muss die Anzahl der Einzelproben erhöht werden. | |
5.1.3. Verpackte Futtermittel
Futtermittel (fest und flüssig) können in Beuteln, Säcken, Dosen, Fässern usw. verpackt sein, die in der Tabelle als Einheiten bezeichnet werden. Große Einheiten (≥ 500 kg bzw. l) sind nach den Vorschriften für lose Futtermittel (siehe Nummern 5.1.1 und 5.1.2) zu beproben.
| Teilpartie | Mindestanzahl der Einheiten, aus denen (mindestens) eine Einzelprobe zu entnehmen ist * |
| 1 bis 20 Einheiten | 1 Einheit ** |
| 21 bis 150 Einheiten | 3 Einheiten ** |
| 151 bis 400 Einheiten | 5 Einheiten ** |
| > 400 Einheiten | 1/4 der √-Anzahl von Einheiten, aus der die Teilpartie besteht ***, bis zu 40 Einheiten |
| *) Kann durch das Öffnen einer Einheit die Analyse beeinträchtigt werden (z.B. leicht verderbliches Nassfutter), so gilt die ungeöffnete Einheit als Einzelprobe.
**) Bei Einheiten, deren Inhalt höchstens 1 kg bzw. 1 l beträgt, gilt der Inhalt einer Originaleinheit als Einzelprobe. ***) Wenn sich hierbei eine Bruchzahl ergibt, ist diese auf die nächsthöhere ganze Zahl aufzurunden. |
|
5.1.4. Futterblöcke und Lecksteine
Zu beproben ist mindestens ein Futterblock oder Leckstein je Teilpartie zu 25 Einheiten, die Höchstzahl beträgt vier Futterblöcke oder Lecksteine.
Bei Futterblöcken oder Lecksteinen mit einem Gewicht von jeweils maximal 1 kg gilt der Inhalt eines Futterblocks oder Lecksteins als Einzelprobe.
5.1.5. Raufutter/Grünfutter
| Teilpartie | Mindestanzahl der Einzelproben * |
| ≤ 5 t | 5 |
| > 5 t | √ 5-mal die Anzahl von Tonnen, aus der die Teilpartie |
| besteht **, bis zu 40 Einzelproben | |
| *) In bestimmten Fällen (z.B. bei Silage) können die vorgeschriebenen Einzelproben nicht ohne eine unannehmbare Beschädigung der Partie entnommen werden. In diesen Fällen ist ein alternatives Probenahmeverfahren zulässig, und entsprechende Leitlinien für die Beprobung solcher Partien werden vor dem Geltungsbeginn dieser Verordnung erarbeitet.
**) Wenn sich hierbei eine Bruchzahl ergibt, ist diese auf die nächsthöhere ganze Zahl aufzurunden. |
|
5.2 Quantitative Anforderungen an Einzelproben bezüglich der Untersuchung auf Bestandteile oder Stoffe, die ungleichmäßig im Futtermittel verteilt sein können
Diese quantitativen Anforderungen an Einzelproben gelten in folgenden Fällen:
Hat die Untersuchungsbehörde den starken Verdacht, dass eine solche ungleichmäßige Verteilung auch im Fall einer Kreuzkontamination durch einen Bestandteil oder einen Stoff in einem Futtermittel-Ausgangserzeugnis auftritt, so können die in der nachstehenden Tabelle festgelegten quantitativen Anforderungen gestellt werden.
| Teilpartie | Mindestanzahl der Einzelproben |
| < 80 t | Siehe quantitative Anforderungen in Nummer 5.1. Die Anzahl der zu entnehmenden Einzelproben ist mit dem Faktor 2,5 zu multiplizieren. |
| ≥ 80 t | 100 |
5.3. Quantitative Anforderungen an Einzelproben bei sehr großen Partien
Bei großen Teilpartien (> 500 t) ist die Anzahl der zu entnehmenden Einzelproben wie folgt zu festzulegen: 40 Einzelproben + √-Tonnen für die Untersuchung auf Stoffe oder Erzeugnisse, die im gesamten Futtermittel gleichmäßig verteilt sind, oder 100 Einzelproben + √-Tonnen für die Untersuchung auf Bestandteile oder Stoffe, die ungleichmäßig in Futtermittel-Ausgangserzeugnissen verteilt sein können.
6. Quantitative Anforderungen an Sammelproben
| Je Teilpartie ist eine einzelne Sammelprobe erforderlich. | ||
| Art des Futtermittels | Mindestgröße der Sammelprobe *, ** | |
| 6.1. | Lose Futtermittel | 4 kg |
| 6.2. | Verpackte Futtermittel | 4 kg *** |
| 6.3. | Flüssige oder halbflüssige Futtermittel | 4 l |
| 6.4. | Futterblöcke oder Lecksteine | |
| 6.4.1. | mit einem Einzelgewicht von mehr als 1 kg | 4 kg |
| 6.4.2. | mit einem Einzelgewicht von höchstens 1 kg | Gewicht von 4 Originalblöcken oder -steinen |
| 6.5. | Raufutter/Grünfutter | 4 kg **** |
| *) Ist das beprobte Futtermittel hochwertig, kann eine kleinere Menge als Sammelprobe gewählt werden, sofern dies im Probenahmebericht beschrieben und dokumentiert wird.
**) Gemäß der Verordnung (EU) Nr. 619/2011 der Kommission vom 24. Juni 2011 zur Festlegung der Probenahme- und Analyseverfahren für die amtliche Untersuchung von Futtermitteln im Hinblick auf genetisch veränderte Ausgangserzeugnisse, für die ein Zulassungsverfahren anhängig ist oder deren Zulassung abläuft (ABl. Nr. L 166 vom 25.06.2011 S. 9) muss die Sammelprobe für die Untersuchung auf genetisch veränderte Ausgangserzeugnisse mindestens 35.000 Samen/Körner enthalten. Dies bedeutet, dass die Größe der Sammelprobe bei Mais mindestens 10,5 kg und bei Sojabohnen 7 kg betragen muss. Bei anderen Samen und Körnern wie Gerste, Hirse, Hafer, Reis, Roggen, Weizen und Raps entspricht die Größe der Sammelprobe von 4 kg mehr als 35.000 Samen/Körnern. ***) Bei verpackten Futtermitteln kann es ebenfalls vorkommen, dass - abhängig von der Größe der einzelnen Einheiten - die für die Sammelprobe vorgeschriebene Größe von 4 kg nicht erreicht werden kann. ****) Handelt es sich um Raufutter oder Grünfutter mit geringer relativer Dichte (z.B. Heu, Stroh), sollte die Probengröße mindestens 1 kg betragen. |
||
7. Quantitative Anforderungen an Endproben
Endproben
Mindestens eine Endprobe muss analysiert werden. Die Menge der zur Analyse bestimmten Endproben darf nicht unter den nachstehend festgelegten Mindestmengen liegen:
| Feste Futtermittel | 500 g *, **, *** |
| Flüssige oder halbflüssige Futtermittel | 500 ml * |
| *) Gemäß der Verordnung (EU) Nr. 619/2011 muss die Endprobe für die Untersuchung auf genetisch veränderte Ausgangserzeugnisse mindestens 10.000 Samen/Körner enthalten. Dies bedeutet, dass die Größe der Endprobe bei Mais mindestens 3.000 g und bei Sojabohnen 2.000 g betragen muss. Bei anderen Samen und Körnern wie Gerste, Hirse, Hafer, Reis, Roggen, Weizen und Raps entspricht die Größe der Endprobe von 500 g mehr als 10.000 Samen/Körnern.
**) Liegt die Größe der Sammelprobe erheblich unter 4 kg bzw. 4 l (siehe Fußnoten in Nummer 6), kann als Endprobe auch eine geringere Menge entnommen werden, sofern dies im Probenahmebericht beschrieben und dokumentiert wird. ***) Bei der Beprobung von Hülsenfrüchten, Getreidekörnern und Schalenfrüchten zur Bestimmung der Pflanzenschutzmittelrückstände muss gemäß der Richtlinie 2002/63/EG der Kommission (ABl. Nr. L 187 vom 16.07.2002 S. 30) die Mindestgröße der Endprobe 1 kg betragen. |
|
8. Probenahmeverfahren für sehr grosse Partien oder Partien, die so gelagert oder befördert werden, dass eine Beprobung der gesamten Partie nicht Praktikabel ist
8.1. Allgemeine Grundsätze
Falls es die Art der Beförderung oder Lagerung einer Partie nicht gestattet, Einzelproben aus der gesamten Partie zu entnehmen, sollte die Beprobung solcher Partien vorzugsweise dann erfolgen, wenn sich die Partie im Fluss befindet.
Falls es sich um Großlager für Futtermittel handelt, sollten die Unternehmer dazu angehalten werden, Einrichtungen im Lager zu installieren, die eine (automatische) Beprobung der gesamten gelagerten Partie ermöglichen.
Im Fall der Anwendung der Probenahmeverfahren nach Kapitel 8 wird der Futtermittelunternehmer oder sein Vertreter über das Probenahmeverfahren informiert. Wird dieses Probenahmeverfahren von dem Futtermittelunternehmer oder seinem Vertreter in Frage gestellt, so muss er es der zuständigen Behörde auf eigene Kosten ermöglichen, die gesamte Partie zu beproben.
8.2. Große Partien, die per Schiff befördert werden
8.2.1. Dynamische Beprobung großer Partien, die per Schiff befördert werden
Die Beprobung großer Partien auf Schiffen ist vorzugsweise durchzuführen, wenn sich das Erzeugnis im Fluss befindet (dynamische Probenahme).
Die Probenahme hat je Laderaum (physisch abtrennbare Einheit) zu erfolgen. Die Laderäume werden allerdings nacheinander geleert, so dass die ursprüngliche physische Trennung nach der Weiterbeförderung in die Lagereinrichtungen nicht mehr besteht. Die Probenahme kann daher unter Bezug auf die ursprüngliche physische Trennung oder auf die Trennung nach der Beförderung in die Lagereinrichtungen erfolgen.
Das Löschen einer Schiffsladung kann mehrere Tage in Anspruch nehmen. In der Regel muss die Beprobung in regelmäßigen Abständen während der gesamten Dauer des Löschvorgangs erfolgen. Es ist jedoch nicht immer praktikabel oder sinnvoll, dass sich ein amtlicher Inspektor während des gesamten Löschvorgangs für die Probenahme vor Ort aufhält. Daher ist die Beprobung eines Teils (einer Teilpartie) der gesamten Partie zulässig. Die Anzahl der Einzelproben wird unter Berücksichtigung des Umfangs der Teilpartie festgelegt.
Wird ein Teil einer Futtermittelpartie derselben Gruppe oder Bezeichnung beprobt und erfüllt diese Teilpartie nachweislich nicht die EU-Anforderungen, so muss davon ausgegangen werden, dass das Ergebnis der Beprobung für die gesamte Futtermittelpartie gilt, es sei denn, eine eingehende Bewertung erbringt keinen Nachweis darüber, dass die restliche Partie den EU-Anforderungen nicht genügt.
Auch wenn die amtliche Probe automatisch entnommen wird, muss ein Inspektor anwesend sein. Erfolgt die automatische Probenahme jedoch anhand voreingestellter Parameter, die während der Probenahme nicht verändert werden können, und werden die Einzelproben in einem verplombten Behälter gesammelt, was einen möglichen Betrug ausschließt, so ist die Anwesenheit eines Inspektors nur zu Beginn der Probenahme, bei jedem Wechsel des Probenbehälters und am Ende der Probenahme erforderlich.
8.2.2. Beprobung von Partien, die per Schiff befördert werden, durch statische Probenahme
Bei einer statischen Probenahme ist dasselbe Verfahren anzuwenden wie bei Lagereinrichtigungen (Silos), die von oben zugänglich sind (siehe Nummer 8.4.1).
Die Probenahme muss am zugänglichen Teil der Partie/des Laderaums erfolgen (von oben). Die Anzahl der Einzelproben wird unter Berücksichtigung des Umfangs der Teilpartie festgelegt. Wird ein Teil einer Futtermittelpartie derselben Gruppe oder Bezeichnung beprobt und erfüllt diese Teilpartie nachweislich nicht die EU-Anforderungen, so muss davon ausgegangen werden, dass das Ergebnis der Beprobung für die gesamte Futtermittelpartie gilt, es sei denn, eine eingehende Bewertung erbringt keinen Nachweis darüber, dass die restliche Partie den EU-Anforderungen nicht genügt.
8.3. Beprobung großer Partien in Lagern
Die Probenahme muss am zugänglichen Teil der Partie erfolgen. Die Anzahl der Einzelproben wird unter Berücksichtigung des Umfangs der Teilpartie festgelegt. Wird ein Teil einer Futtermittelpartie derselben Gruppe oder Bezeichnung beprobt und erfüllt diese Teilpartie nachweislich nicht die EU-Anforderungen, so muss davon ausgegangen werden, dass das Ergebnis der Beprobung für die gesamte Futtermittelpartie gilt, es sei denn, eine eingehende Bewertung erbringt keinen Nachweis darüber, dass die restliche Partie den EU-Anforderungen nicht genügt.
8.4. Beprobung von Lagereinrichtungen (Silos)
8.4.1. Beprobung von Silos mit (leichtem) Zugang von oben
Die Probenahme muss am zugänglichen Teil der Partie erfolgen. Die Anzahl der Einzelproben wird unter Berücksichtigung des Umfangs der Teilpartie festgelegt. Wird ein Teil einer Futtermittelpartie derselben Gruppe oder Bezeichnung beprobt und erfüllt diese Teilpartie nachweislich nicht die EU-Anforderungen, so muss davon
ausgegangen werden, dass das Ergebnis der Beprobung für die gesamte Futtermittelpartie gilt, es sei denn, eine eingehende Bewertung erbringt keinen Nachweis darüber, dass die restliche Partie den EU-Anforderungen nicht genügt.
8.4.2. Beprobung von Silos ohne Zugang von oben (geschlossene Silos)
8.4.2.1. Silos ohne Zugang von oben (geschlossene Silos) mit einer Größe über 100 Tonnen
In solchen Silos gelagerte Futtermittel können nicht statisch beprobt werden. Wenn das im Silo gelagerte Futtermittel beprobt werden muss und keine Möglichkeit besteht, die Sendung zu bewegen, ist eine Vereinbarung mit dem Unternehmer dahin gehend zu treffen, dass dieser den Inspektor darüber informiert, wann das Silo geleert wird, damit eine Probenahme erfolgen kann, wenn sich das Futtermittel im Fluss befindet.
8.4.2.2. Silos ohne Zugang von oben (geschlossene Silos) mit einer Größe unter 100 Tonnen
Im Rahmen des Probenahmeverfahrens ist eine Menge von 50 bis 100 kg in einen Behälter abzufüllen und anschließend zu beproben. Die Größe der Sammelprobe entspricht der gesamten Partie, und die Anzahl der Einzelproben muss im Verhältnis zu der Menge stehen, die zur Probenahme aus dem Silo in einen Behälter abgefüllt wird. Wird ein Teil einer Futtermittelpartie derselben Gruppe oder Bezeichnung beprobt und erfüllt diese Teilpartie nachweislich nicht die EU-Anforderungen, so muss davon ausgegangen werden, dass das Ergebnis der Beprobung für die gesamte Futtermittelpartie gilt, es sei denn, eine eingehende Bewertung erbringt keinen Nachweis darüber, dass die restliche Partie den EU-Anforderungen nicht genügt.
8.5. Beprobung loser Futtermittel in großen geschlossenen Containern
Solche Partien können häufig erst nach dem Entladen beprobt werden. In bestimmten Fällen ist das Entladen am Einfuhrort oder am Kontrollpunkt nicht möglich, weshalb die Probenahme erfolgen sollte, wenn die betreffenden Container entladen sind.
9. Anweisungen für die Entnahme, Vorbereitung und Verpackung der Proben
9.1. Allgemeines
Die Proben müssen so schnell wie möglich entnommen und vorbereitet werden, wobei die nötigen Vorkehrungen zu treffen sind, um sicherzustellen, dass das Erzeugnis weder verändert noch verunreinigt wird. Die für die Probenahme bestimmten Geräte, Flächen und Behälter müssen sauber und trocken sein.
9.2. Einzelproben
Die Einzelproben sind nach dem Zufallsprinzip aus der gesamten Teilpartie zu entnehmen. Ihre Größe muss ungefähr gleich sein.
Die Größe der Einzelprobe muss mindestens 100 g bzw. 25 g bei Raufutter oder Grünfutter mit geringem spezifischem Gewicht betragen.
Sind nach dem Probenahmeverfahren gemäß Nummer 8 weniger als 40 Einzelproben zu entnehmen, muss die Größe der Einzelprobe im Verhältnis zur erforderlichen Größe der Sammelprobe festgelegt werden (siehe Nummer 6).
Bei der Beprobung kleiner Partien verpackter Futtermittel, bei der entsprechend den quantitativen Anforderungen eine begrenzte Anzahl von Einzelproben zu entnehmen ist, umfasst eine Einzelprobe den Inhalt einer Originaleinheit, der 1 kg bzw. 1 Liter nicht überschreitet.
Bei der Beprobung verpackter Futtermittel, die aus kleinen Einheiten bestehen (z.B. < 250 g) ist die Größe der Einzelprobe von der Größe der Einheit abhängig.
9.2.1. Lose Futtermittel
Die Probenahme kann gegebenenfalls auch bei einer Teilpartie erfolgen, die sich in Bewegung (Einladen oder Entladen) befindet.
9.2.2. Verpackte Futtermittel
Nach Auswahl der erforderlichen Anzahl der zu beprobenden Einheiten gemäß Kapitel 5 wird aus jeder dieser Einheiten ein Teil des Inhalts mit einem Probenahmestab oder einer Schaufel entnommen. Gegebenenfalls sind die Proben zu entnehmen, nachdem die Einheiten getrennt entleert worden sind.
9.2.3. Homogene oder homogenisierbare flüssige oder halbflüssige Futtermittel
Nach Auswahl der erforderlichen Anzahl der zu beprobenden Einheiten gemäß Kapitel 5 wird der Inhalt falls nötig homogenisiert und aus jeder Einheit eine Menge entnommen.
Die Einzelproben können auch beim Ablassen des Inhalts entnommen werden.
9.2.4. Nicht homogenisierbare flüssige oder halbflüssige Futtermittel
Nach Auswahl der erforderlichen Anzahl der zu beprobenden Einheiten gemäß Kapitel 5 werden in unterschiedlicher Höhe Proben entnommen.
Die Probenahme kann auch beim Ablassen des Inhalts erfolgen, wobei jedoch die ersten Fraktionen zu verwerfen sind.
In beiden Fällen muss das Gesamtvolumen mindestens 10 l betragen.
9.2.5. Futterblöcke und Lecksteine
Nach Auswahl der erforderlichen Anzahl der zu beprobenden Blöcke oder Steine gemäß Kapitel 5 kann jedem Block oder Stein ein Teil entnommen werden. Besteht die Vermutung, dass es sich um einen nicht homogenen Block bzw. Stein handelt, so kann der ganze Block bzw. Stein als Probe genommen werden.
Bei Futterblöcken oder Lecksteinen mit einem Gewicht von jeweils maximal 1 kg gilt der Inhalt eines Futterblocks oder Lecksteins als Einzelprobe.
9.3. Vorbereitung der Sammelproben
Die Einzelproben werden gemischt und zu einer einzigen Sammelprobe zusammengestellt.
9.4. Vorbereitung der Endproben
Das Material in der Sammelprobe ist sorgfältig zu mischen 1.
9.5. Verpackung der Proben
Die Behälter oder Packungen sind so zu verplomben und zu kennzeichnen, dass sie nicht ohne Beschädigung der Plombe geöffnet werden können. Hierbei muss die gesamte Kennzeichnung von der Plombe erfasst werden.
9.6. Versendung der Proben an das Labor
Die Probe ist - zusammen mit den untersuchungsrelevanten Angaben - so schnell wie möglich an das benannte Analyselabor zu versenden.
10. Probenahmeprotokoll
Für jede Probe ist ein Probenahmeprotokoll zu erstellen, aus dem die Identität und der Umfang der Teilpartie eindeutig hervorgehen.
Im Protokoll wird außerdem jede Abweichung von den in dieser Verordnung festgelegten Probenahmeverfahren vermerkt.
Das Protokoll wird dem amtlichen Kontrolllabor sowie dem Futtermittelunternehmer und/oder dem vom Futtermittelunternehmer benannten Labor zur Verfügung gestellt.
_________
1) Klumpen sind zu zerdrücken (sie werden gegebenenfalls vom übrigen Material getrennt und der Probe anschließend wieder untergemischt).
2) Außer bei Raufutter oder Grünfutter mit geringem spezifischem Gewicht.
3) Außer bei Raufutter oder Grünfutter mit geringem spezifischem Gewicht.
| Allgemeine Bestimmungen hinsichtlich der Methoden zur Analyse von Futtermitteln | Anhang II13 |
A. Vorbereitung der Proben zur Analyse
1. Zweck
Die nachfolgend beschriebenen Verfahren betreffen die Vorbereitung der zur Analyse bestimmten Proben, die nach der Probenahme gemäß den Bestimmungen in Anhang I an die Kontrolllabors versandt wurden.
Die Vorbereitung der Laborproben muss so erfolgen, dass die in den Analysemethoden vorgesehenen Einwaagen homogen und repräsentativ für die Endproben sind.
2. Vorsichtsmaßnahmen
Die Wahl des Verfahrens für die Probenvorbereitung hängt von der Art der anzuwendenden Analysemethode und den zu untersuchenden Bestandteilen oder Stoffen ab. Daher ist es äußerst wichtig, sicherzustellen, dass das angewandte Probenvorbereitungsverfahren für die jeweilige Analysemethode und für die zu untersuchenden Bestandteile oder Stoffe geeignet ist.
Alle notwendigen Schritte sind so durchzuführen, dass eine Verunreinigung der Probe und eine Veränderung ihrer Zusammensetzung weitestmöglich vermieden werden.
Das Zerkleinern, Mischen und Sieben muss unverzüglich erfolgen, wobei die Probe möglichst wenig Luft und Licht ausgesetzt werden darf. Die Verwendung von Mühlen oder Zerkleinerungsgeräten, die zu einer merklichen Erwärmung der Probe führen könnten, ist nicht zulässig.
Für besonders hitzeempfindliche Futtermittel wird manuelles Zerkleinern empfohlen. Es ist außerdem darauf zu achten, dass die verwendeten Geräte keine Kontaminationsquelle bilden.
Kann die Probe nicht ohne eine signifikante Veränderung ihres Feuchtigkeitsgehalts vorbereitet werden, so ist dieser vor und nach der Vorbereitung gemäß der in Anhang III Teil A festgelegten Methode zu bestimmen.
3. Verfahren
3.1 Allgemeines Verfahren
Das Testaliquot ist der Endprobe zu entnehmen. Das Kegeln und das Vierteilungsverfahren werden nicht empfohlen, da dies mit einer hohen Teilungsfehlerquote bei den Testaliquoten einhergehen kann.
3.1.1. Futtermittel, die direkt gemahlen werden können
3.1.2. Futtermittel, die nach Trocknung gemahlen werden können
3.1.3. Flüssige oder halbflüssige Futtermittel
3.1.4. Andere Futtermittel
3.2 Besonderes Verfahren bei Sichtprüfungen oder mikroskopischen Untersuchungen oder im Fall einer Homogenisierung der vollständigen Sammelprobe
4. Lagerung der Proben
Die Proben müssen bei einer Temperatur gelagert werden, die ihre Zusammensetzung nicht beeinflusst. Proben, die zur Analyse von Vitaminen oder besonders lichtempfindlichen Stoffen bestimmt sind, sind so zu lagern, dass die Probe nicht durch Licht beeinträchtigt wird.
B. Bestimmungen über in Analyseverfahren verwendete Reagenzien und Geräte
C. Anwendung von Analysemethoden und Formulierung der Ergebnisse
1. Extraktionsverfahren
Einige Methoden geben ein spezifisches Extraktionsverfahren vor. Generell können andere Extraktionsverfahren als das in der Methode genannte Verfahren angewandt werden, und zwar unter der Bedingung, dass das angewandte Extraktionsverfahren für die analysierte Matrix eine vergleichbare Extraktionseffizienz aufweist wie das in der Methode genannte Verfahren.
2. Cleanup-Verfahren
Einige Methoden geben ein spezifisches Cleanup-Verfahren vor. Generell können andere Cleanup-Verfahren als das in der Methode genannte Verfahren angewandt werden, und zwar unter der Bedingung, dass das angewandte Cleanup-Verfahren für die analysierte Matrix zu vergleichbaren Analyseergebnissen führt wie das in der Methode genannte Verfahren.
3. Anzahl der Bestimmungen
Bei der Analyse auf unerwünschte Stoffe sind - sofern das Ergebnis der ersten Bestimmung deutlich (> 50 %) unter dem zu kontrollierenden Sollwert liegt - keine weiteren Bestimmungen erforderlich, vorausgesetzt, dass die geeigneten Qualitätsverfahren angewandt werden. In anderen Fällen ist eine Zweitanalyse (Zweitbestimmung) erforderlich, um eine interne Kreuzkontamination oder eine versehentliche Vermischung der Proben auszuschließen. Anhand des Mittelwerts der beiden Bestimmungen wird - unter Berücksichtigung der Messunsicherheit - die Einhaltung der Höchstgehalte überprüft.
Bei der Kontrolle des angegebenen Gehalts an einem Stoff oder einer Zutat sind - sofern das Ergebnis der Erstbestimmung den angegebenen Gehalt bestätigt, d. h., wenn das Analyseergebnis innerhalb des akzeptablen Toleranzbereichs für den angegebenen Gehalt liegt - keine weiteren Bestimmungen erforderlich, vorausgesetzt, dass die geeigneten Qualitätsverfahren angewandt werden. In anderen Fällen ist eine Zweitanalyse (Zweitbestimmung) erforderlich, um eine interne Kreuzkontamination oder eine versehentliche Vermischung der Proben auszuschließen. Anhand des Mittelwerts der beiden Bestimmungen wird - unter Berücksichtigung der Messunsicherheit - die Einhaltung der Höchstgehalte überprüft.
In einigen Fällen ist der Toleranzbereich in Rechtsvorschriften definiert, beispielsweise in der Verordnung (EG) Nr. des Europäischen Parlaments und des Rates vom 13. Juli 2009 über das Inverkehrbringen und die Verwendung von Futtermitteln, zur Änderung der Verordnung (EG) Nr. 1831/2003 des Europäischen Parlaments und des Rates und zur Aufhebung der Richtlinien 79/373/EWG des Rates, 80/511/EWG der Kommission, 82/471/EWG des Rates, 83/228/EWG des Rates, 93/74/EWG des Rates, 93/113/EG des Rates und 96/25/EG des Rates und der Entscheidung 2004/217/EG der Kommission 1.
4. Berichterstattung über die angewandte Analysemethode
Im Analysebericht ist anzugeben, welche Analysemethode angewandt wurde.
5. Bericht über die Analyseergebnisse
Das Ergebnis ist gemäß den Anweisungen in der Analysemethode mit einer angemessenen Zahl an signifikanten Ziffern anzugeben und, falls nötig, auf den Feuchtigkeitsgehalt der Endprobe vor deren Vorbereitung umzurechnen.
6. Messunsicherheit und Wiederfindungsrate bei der Analyse auf unerwünschte Stoffe
Hinsichtlich unerwünschter Stoffe im Sinne der Richtlinie 2002/32/EG erfüllt ein zur Verfütterung bestimmtes Erzeugnis die Bestimmung bezüglich des festgelegten Höchstgehalts nicht, wenn das Analyseergebnis, bezogen auf ein Futtermittel mit einem Feuchtigkeitsgehalt von 12 %, unter Berücksichtigung der erweiterten Messunsicherheit und der Berichtigung um die Wiederfindungsrate den Höchstgehalt überschreitet. Zur Beurteilung der Einhaltung der Höchstgehalte wird die gemessene Konzentration - nach Berichtigung um die Wiederfindungsrate und nach Abzug der erweiterten Messunsicherheit - herangezogen. Dieses Verfahren wird nur dann angewandt, wenn die Analysemethode die Schätzung der Messunsicherheit und die Berichtigung um die Wiederfindungsrate ermöglicht (z.B. nicht möglich bei mikroskopischer Analyse).
Das Analyseergebnis ist wie folgt festzuhalten (soweit die angewandte Analysemethode die Schätzung der Messunsicherheit und der Wiederfindungsrate ermöglicht):
Liegt jedoch das Analyseergebnis deutlich (> 50 %) unter dem zu kontrollierenden Sollwert - und unter der Bedingung, dass die geeigneten Qualitätsverfahren angewandt werden und die Analyse lediglich dem Zweck der Überprüfung der Einhaltung der Rechtsvorschriften dient -, können die Analyseergebnisse ohne Berichtigung um die Wiederfindungsrate angegeben werden, und die Angabe der Wiederfindungsrate und der Messunsicherheit kann in diesen Fällen entfallen.
________
1) ABl. Nr. L 229 vom 01.09.2009 S. 1.
| Analysemethoden zur Untersuchung der Zusammensetzung von Futtermittelausgangserzeugnissen und Mischfuttermitteln | Anhang III |
A. Bestimmung des Feuchtigkeitsgehalts
1. Zweck und Anwendungsbereich
Diese Methode erlaubt die Bestimmung des Feuchtigkeitsgehalts von Futtermitteln. Bei Futtermitteln, die flüchtige Stoffe wie z.B. organische Säuren enthalten, ist zu beachten, dass zusammen mit dem Feuchtigkeitsgehalt auch bedeutende Mengen an flüchtigen Stoffen bestimmt werden.
Sie betrifft nicht die Untersuchung von Milcherzeugnissen als Futtermittel-Ausgangserzeugnisse, die Untersuchung von Mineralstoffen und Mischungen, die überwiegend aus Mineralstoffen bestehen, die Untersuchung von tierischen und pflanzlichen Fetten und Ölen oder die Untersuchung von Ölsaaten und Ölfrüchten.
2. Prinzip
Die Probe wird unter definierten Bedingungen getrocknet, die von der Art des Futtermittels abhängen. Der Gewichtsverlust wird durch Wiegen bestimmt. Bei festen Futtermitteln mit einem hohen Feuchtigkeitsgehalt ist eine zusätzliche Vortrocknung erforderlich.
3. Geräte
3.1 Zerkleinerungsgerät aus einem Material, das keine Feuchtigkeit absorbiert, leicht zu reinigen ist, eine schnelle und gleichmäßige Zerkleinerung ermöglicht, ohne eine merkliche Erwärmung hervorzurufen, das so weit wie möglich den Kontakt mit der Außenluft verhindert und den unter 4.1.1 und 4.1.2 gestellten Anforderungen entspricht (z.B. Mikroschlagkreuzmühlen, Mikrozerkleinerer mit Wasserkühlung, zerlegbare Kegelmühlen, langsam laufende Kegelmühlen oder Zahnscheibenmühlen).
3.2 Analysenwaage, Genauigkeit 1 mg.
3.3 Trockene Gefäße aus korrosionsbeständigem Metall oder Glas, mit luftdicht schließenden Deckeln; die Nutzfläche muss eine Verteilung der Probe von ca. 0,3 g/cm2 ermöglichen.
3.4 Elektrisch beheizter, temperaturgeregelter Trockenschrank (± 2 °C), der eine schnelle Regelung der Temperatur gewährleistet und eine gute Lüftung besitzt1.
3.5 Elektrisch beheizter regulierbarer Vakuumtrockenschrank mit einer Ölpumpe, der entweder mit einer Vorrichtung für die Zufuhr warmer und getrockneter Luft oder mit einem Trocknungsmittel (z.B. Calciumoxid) ausgestattet ist.
3.6 Exsikkator mit dicker, perforierter Platte aus Metall oder Porzellan, der ein wirksames Trocknungsmittel enthält.
4. Verfahren
Anmerkung:
Die Verfahren, die in diesem Abschnitt beschrieben werden, müssen unverzüglich nach dem Öffnen der Packungen, die die Proben enthalten, durchgeführt werden. Die Analysen sind mindestens doppelt auszuführen.
4.1 Vorbereitung
4.1.1 Futtermittel, außer den unter 4.1.2 und 4.1.3 genannten
Mindestens 50 g der Probe werden entnommen und erforderlichenfalls unter Vermeidung von Feuchtigkeitsänderungen entsprechend zerkleinert oder geteilt (siehe 6).
4.1.2 Getreide und Grütze
Mindestens 50 g der Probe werden entnommen. Diese Menge wird so gemahlen, dass zumindest 50 % der Teilchen durch ein Sieb mit einer Maschenweite von 0,5 mm passiert werden können und dass beim Passieren durch ein Rundlochsieb mit einem Lochdurchmesser von 1 mm ein Rückstand von höchstens 10 % verbleibt.
4.1.3 Flüssige oder breiige Futtermittel; Futtermittel, die überwiegend aus Ölen und Fetten bestehen
Etwa 25 g der Probe, auf 10 mg genau gewogen, werden entnommen, mit einer entsprechenden, auf 10 mg genau gewogenen Menge wasserfreiem Sand vermischt, bis ein homogenes Produkt entsteht.
4.2 Trocknung
4.2.1 Futtermittel, außer den unter 4.2.2 und 4.2.3 genannten
Ein Gefäß ( 3.3) wird mit seinem Deckel auf 1 mg genau gewogen. Etwa 5 g der Probe werden auf 1 mg genau in das tarierte Gefäß eingewogen und gleichmäßig verteilt. Das Gefäß wird ohne Deckel in den auf 103 °C vorgeheizten Trockenschrank gestellt. Damit die Temperatur des Trockenschranks nicht zu stark abfällt, ist das Gefäß möglichst rasch hineinzustellen. Es wird 4 h lang getrocknet, wobei die Trocknungszeit von dem Zeitpunkt an gerechnet wird, an dem die Temperatur im Trockenschrank erneut 103 °C erreicht hat. Nach Öffnen des Trockenschranks wird das Gefäß sofort mit dem Deckel verschlossen, aus dem Schrank genommen, 30 bis 45 min lang zum Abkühlen in den Exsikkator ( 3.6) gestellt und anschließend auf 1 mg genau gewogen.
Bei Futtermitteln, die überwiegend aus Ölen und Fetten bestehen, wird eine zusätzliche Trocknung von 30 min im Trockenschrank bei 130 °C vorgenommen. Der Unterschied zwischen den beiden Wägeergebnissen darf höchstens 0,1 % Feuchtigkeit betragen.
4.2.2 Getreide, Mehl, Grütze und Grieß
Ein Gefäß ( 3.3) wird mit seinem Deckel auf 0,5 mg genau gewogen. Etwa 5 g der zerkleinerten Probe werden auf 1 mg genau in das tarierte Gefäß eingewogen und gleichmäßig verteilt. Das Gefäß wird ohne Deckel in den auf 130 °C vorgeheizten Trockenschrank gestellt. Damit die Temperatur des Trockenschranks nicht zu stark abfällt, ist das Gefäß möglichst rasch hineinzustellen. Es wird 2 h lang getrocknet, wobei die Trocknungszeit von dem Zeitpunkt an gerechnet wird, an dem die Temperatur im Trockenschrank erneut 130 °C erreicht hat. Nach Öffnen des Trockenschranks wird das Gefäß sofort mit dem Deckel verschlossen, aus dem Schrank genommen, 30 bis 45 min lang zum Abkühlen in den Exsikkator ( 3.6) gestellt und anschließend auf 1 mg genau gewogen.
4.2.3 Mischfuttermittel mit einem Saccharose- oder Lactosegehalt von mehr als 4 %: Futtermittel-Ausgangserzeugnisse wie z.B. Johannisbrotschrot, hydrolysierte Getreideerzeugnisse, Malzkeime, Zuckerrübenschnitzel, Fisch- und Zuckerpresssäfte; Mischfuttermittel mit einem Gehalt an kristallwasserhaltigen Mineralstoffen von mehr als 25 %.
Ein Gefäß ( 3.3) wird mit seinem Deckel auf 0,5 mg genau gewogen. Etwa 5 g der Probe werden auf 1 mg genau in das tarierte Gefäß eingewogen und gleichmäßig verteilt. Das Gefäß wird ohne Deckel in den auf 80 bis 85 °C vorgeheizten Vakuumtrockenschrank ( 3.5) gestellt. Damit die Temperatur des Vakuumtrockenschranks nicht zu stark abfällt, ist das Gefäß möglichst rasch hineinzustellen.
Der Druck wird auf 100 Torr eingestellt. Die Probe wird 4 h lang bei diesem Druck entweder unter Zufuhr von heißer trockener Luft oder mittels eines Trocknungsmittels (etwa 300 g für 20 Proben) getrocknet. Im letzteren Fall wird beim Erreichen des vorgeschriebenen Drucks die Verbindung zur Vakuumpumpe unterbrochen. Die Trocknungszeit wird von dem Zeitpunkt an gerechnet, an dem die Temperatur im Trockenschrank erneut 80 bis 85 °C erreicht hat. Nach Ablauf der Trocknungszeit wird der Vakuumtrockenschrank vorsichtig wieder auf atmosphärischen Druck gebracht. Nach Öffnen des Vakuumtrockenschranks wird das Gefäß sofort mit dem Deckel verschlossen, aus dem Schrank genommen, zum Abkühlen 30 bis 45 min lang in den Exsikkator ( 3.6) gestellt und anschließend auf 1 mg genau gewogen. Es wird im Vakuumtrockenschrank weitere 30 min bei 80 bis 85 °C getrocknet und erneut gewogen. Der Unterschied zwischen den beiden Wägeergebnissen darf höchstens 0,1 % Feuchtigkeit betragen.
4.3 Vortrocknung
4.3.1 Futtermittel, außer den unter 4.3.2 genannten
Bei festen Futtermitteln mit einem hohen Feuchtigkeitsgehalt, deren Zerkleinerung schwierig ist, ist eine Vortrocknung wie folgt vorzunehmen:
Von der nicht zerkleinerten Probe (sofern erforderlich können gepresste oder klumpenhaltige Futtermittel grob zerkleinert werden) werden 50 g auf 10 mg genau in ein geeignetes Gefäß (z.B. eine Aluminiumschale von 20 × 12 cm mit einem Rand von 0,5 cm) eingewogen und in einem Trockenschrank bei einer Temperatur von 60 bis 70 °C getrocknet, bis der Feuchtigkeitsgehalt einen Wert zwischen 8 und 12 % erreicht hat. Anschließend wird das Gefäß aus dem Trockenschrank genommen, im Labor 1 h lang offen abkühlen gelassen und dann auf 10 mg genau gewogen. Im Folgenden wird die Probe unverzüglich, wie unter 4.1.1 angegeben, zerkleinert und je nach Art des Futtermittels entsprechend den Angaben unter 4.2.1 oder 4.2.3 getrocknet.
4.3.2 Getreide
Körner, deren Feuchtigkeitsgehalt höher als 17 % ist, müssen wie folgt vorgetrocknet werden:
Von den nicht gemahlenen Körnern werden 50 g auf 10 mg genau in ein geeignetes Gefäß (z.B. in eine Aluminiumschale von 20 x 12 cm mit einem Rand von 0,5 cm) eingewogen und in einem Trockenschrank bei einer Temperatur von 130 °C 5 bis 7 min getrocknet. Anschließend wird das Gefäß aus dem Trockenschrank genommen, im Labor 2 h lang offen abkühlen gelassen und dann auf 10 mg genau gewogen. Im Folgenden wird die Probe unverzüglich, wie unter 4.1.2 angegeben, gemahlen und entsprechend den Angaben unter 4.2.2 getrocknet.
5. Berechnung der Ergebnisse
Der Feuchtigkeitsgehalt (X) der Probe als Prozentsatz wird nach folgenden Formeln berechnet:
5.1 Trocknung- ohne Vortrocknung
wobei:
m = Anfangsgewicht der Probe in g,
m0 = Gewicht der trockenen Probe in g.
5.2 Trocknung mit Vortrocknung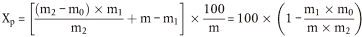
wobei:
m = Anfangsgewicht der Probe in g,
m1 = Gewicht der Probe nach der Vortrocknung in g,
m2 = Gewicht der Probe nach der Zerkleinerung oder dem Zermahlen in g,
m0 = Gewicht der trockenen Probe in g.
5.3 Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 0,2 % Feuchtigkeit (absolut) nicht überschreiten.
6. Bemerkung
Wenn eine Zerkleinerung sich als notwendig erweist und dabei mit einer Änderung des Feuchtigkeitsgehalts des Materials gerechnet werden muss, so sind die Analyseergebnisse, die die Bestandteile des Futtermittels betreffen, auf den Feuchtigkeitsgehalt der Originalprobe umzurechnen.
B. Bestimmung des Feuchtigkeitsgehalts in tierischen und pflanzlichen Fetten und Ölen
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an Wasser und flüchtigen Stoffen in tierischen und pflanzlichen Fetten und Ölen.
2. Prinzip
Die Probe wird bei 103 °C getrocknet, bis kein Gewichtsverlust mehr eintritt (der Gewichtsverlust zwischen 2 aufeinanderfolgenden Wägungen darf 1 mg nicht überschreiten). Der Gewichtsverlust wird durch Wiegen bestimmt.
3. Geräte
3.1 Schale mit flachem Boden, aus korrosionsbeständigem Material, Durchmesser. 8 bis 9 cm, Höhe ca. 3 cm.
3.2 Thermometer mit verstärkter Kugel und Ausdehnungsraum am oberen Ende, von ca. 80 bis mindestens 110 °C graduiert, Länge ca. 10 cm.
3.3 Sandbad oder elektrische Heizplatte.
3.4 Exsikkator, der ein wirksames Trocknungsmittel enthält.
3.5 Analysenwaage.
4. Verfahren
Etwa 20 g der homogenisierten Probe werden auf 1 mg genau in die trockene, gewogene Schale ( 3.1.) eingewogen, die das Thermometer ( 3.2) enthält, und auf dem Sandbad oder der elektrischen Heizplatte ( 3.3) unter ständigem Rühren mit dem Thermometer so erhitzt, dass in ca. 7 min eine Temperatur von 90 °C erreicht wird.
Entsprechend der Häufigkeit, mit der Gasblasen vom Boden der Schale aufsteigen, wird die Heizintensität verringert. Die Temperatur darf 105 °C nicht überschreiten. Unter ständigem Abkratzen des Bodens der Schale wird weiter umgerührt, bis sich keine Blasen mehr bilden.
Um sicherzustellen, dass keine Feuchtigkeit mehr vorhanden ist, wird mehrmals auf 103 °C ± 2 °C erhitzt und zwischen aufeinanderfolgenden Erhitzungen jeweils auf 93 °C gekühlt. Anschließend wird bis zur Raumtemperatur im Exsikkator ( 3.4) abkühlen gelassen und gewogen. Dieses Verfahren ist so oft zu wiederholen, bis der Gewichtsverlust zwischen 2 aufeinanderfolgenden Wägungen 2 mg nicht mehr überschreitet.
Anmerkung:
Eine Gewichtserhöhung der Probe nach wiederholter Erwärmung zeigt eine Oxidation des Fettes an. In diesem Fall wird der Berechnung das Ergebnis der Wägung zugrunde gelegt, die unmittelbar vor dem Auftreten der Gewichtszunahme ausgeführt wurde.
5. Berechnung der Ergebnisse
Der Feuchtigkeitsgehalt (X) der Probe als Prozentsatz wird nach folgender Formel berechnet:
wobei:
m = Probeneinwaage in g,
m1 = Gewicht der Schale mit Inhalt in g vor dem Erhitzen,
m2 = Gewicht der Schale mit Inhalt in g nach dem Erhitzen.
Ergebnisse unter 0,05 % sollten mit der Bezeichnung "weniger als 0,05 %" angegeben werden.
Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 0,05 % Feuchtigkeit (absolut) nicht überschreiten.
C. Bestimmung des Rohproteingehalts
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Berechnung des Rohproteingehalts von Futtermitteln anhand des nach Kjeldahl bestimmten Stickstoffgehalts.
2. Prinzip
Die Probe wird durch Schwefelsäure in Anwesenheit eines Katalysators aufgeschlossen. Der saure Aufschluss wird durch eine Natriumhydroxidlösung alkalisiert. Das Ammoniak wird durch Destillation abgetrennt und in einer definierten Menge Schwefelsäure aufgefangen, deren Überschuss durch eine Standard-Natriumhydroxidlösung titriert wird.
Alternativ wird das freigesetzte Ammoniak in überschüssiger Borsäurelösung destilliert, gefolgt von einer Titration mit Salz- oder Schwefelsäurelösung.
3. Reagenzien
3.1 Kaliumsulfat.
3.2 Katalysator: Kupfer(II)-oxid, CuO, oder Kupfer(II)-sulfatpentahydrat, CuSO4 5H2O.
3.3 Zinkgranulat.
3.4 Schwefelsäure, ρ20 = 1,84 g/ml.
3.5 Schwefelsäure, volumetrische Standardlösung, c(H2SO4) = 0,25 mol/l.
3.6 Schwefelsäure, volumetrische Standardlösung, c(H2SO4) = 0,10 mol/l.
3.7 Schwefelsäure, volumetrische Standardlösung, c(H2SO4) = 0,05 mol/l.
3.8 Methylrot-Indikator; 300 mg Methylrot werden in 100 ml Ethanol (σ = 95 bis 96 %) gelöst.
3.9 Natriumhydroxidlösung (Verwendung technischer Qualität möglich), β = 40 g/100 ml (40 %).
3.10 Natriumhydroxid, volumetrische Standardlösung, c(NaOH) = 0,25 mol/l.
3.11 Natriumhydroxid, volumetrische Standardlösung, c(NaOH) = 0,10 mol/l.
3.12 Bimssteinkörner, mit Salzsäure gewaschen und geglüht.
3.13 Acetanilid (Schmelzpunkt = 114 °C, N-Gehalt = 10,36 %).
3.14 Saccharose (stickstofffrei).
3.15 Borsäure (H3BO3).
3.16 Methylrot-Indikatorlösung: 100 mg Methylrot werden in 100 ml Ethanol oder Methanol gelöst.
3.17 Bromkresolgrünlösung: 100 mg Bromkresolgrün werden in 100 ml Ethanol oder Methanol gelöst.
3.18 Borsäurelösung (10 bis 40 g/l in Abhängigkeit vom eingesetzten Gerät)
Bei einer kolorimetrischen Endpunktbestimmung sind der Borsäurelösung Methylrot- und Bromkresolindikatoren zuzufügen. Wird 1 l Borsäurelösung zubereitet, sind vor dem Auffüllen zur Marke 7 ml Methylrot-Indikatorlösung ( 3.16) und 10 ml Bromkresolgrünlösung ( 3.17) zuzufügen.
In Abhängigkeit von dem verwendeten Wasser kann sich der pH-Wert von einer Borsäurelösung zur anderen unterscheiden. Häufig ist eine Einstellung des pH-Werts mithilfe einer geringen Menge Alkali erforderlich, um eine positive Blindprobe zu erhalten.
Anmerkung:
Eine Zugabe von rund 3 ml NaOH ( 3.11) zu 1 l Borsäure von 10 g/l führt in der Regel zu guten Einstellungen. Die Lösung ist bei Raumtemperatur aufzubewahren und während der Aufbewahrung vor Lichteinfall und Ammoniakdämpfen zu schützen.
3.19 Salzsäure, volumetrische Standardlösung, c(HCl) = 0,10 mol/l.
Anmerkung:
Andere Konzentrationen volumetrischer Lösungen ( 3.5, 3.6, 3.7, 3.10, 3.11 und 3.19) können verwendet werden, sofern die Berechnungen entsprechend berichtigt werden. Die Konzentrationen sind stets auf 4 Dezimalstellen genau anzugeben.
4. Geräte
Für Aufschluss, Destillation und Titration nach dem Kjeldahl-Verfahren geeignete Geräte.
5. Verfahren
5.1 Aufschluss
Von der Probe wird 1 g auf 0,001 g genau gewogen und in den Kolben des Aufschlussgeräts gegeben. Anschließend werden 15 g Kaliumsulfat ( 3.1), eine geeignete Katalysatormenge ( 3.2) (0,3 bis 0,4 g Kupfer(II)-oxid oder 0,9 bis 1,2 g Kupfer(II)-sulfatpentahydrat), 25 ml Schwefelsäure ( 3.4) und ggf. einige Bimssteinkörnchen ( 3.12) zugesetzt und vermischt.
Der Kolben wird, wenn notwendig, unter regelmäßigem Schwenken zunächst mäßig erhitzt, bis die Substanz verkohlt ist und das Schäumen aufgehört hat. Dann wird die Flüssigkeit stärker erhitzt und gleichmäßig am Sieden gehalten. Bei korrekter Erhitzung kondensiert die siedende Säure auf der Kolbenwand. Ein Überhitzen der Kolbenwände und Ansetzen organischer Partikel ist zu vermeiden.
Sobald die Lösung klar ist und sich hellgrün färbt, wird sie noch weitere 2 h am Sieden gehalten und danach abkühlen gelassen.
5.2 Destillation
Es wird vorsichtig so viel Wasser zugegeben, dass die Sulfate vollständig gelöst werden. Nach dem Abkühlen werden ggf. einige Körner Zinkgranulat ( 3.3) zugesetzt. Es wird entweder gemäß 5.2.1 oder 5.2.2 weiterverfahren.
5.2.1 Destillation von Schwefelsäure
In den Auffangkolben des Destillationsapparats werden je nach dem zu erwartenden Stickstoffgehalt genau 25 ml Schwefelsäure ( 3.5 oder 3.7) gebracht und einige Tropfen Methylrot-Indikator ( 3.8) hinzugefügt.
Der Aufschlusskolben wird mit dem Kühler des Destillationsapparats verbunden und das Ende des Kühlers mindestens 1 cm tief in die Flüssigkeit des Auffangkolbens gesenkt (siehe Bemerkung 8.3). Dann werden 100 ml Natriumhydroxidlösung ( 3.9) langsam in den Aufschlusskolben eingefüllt, wobei kein Ammoniak entweichen darf (siehe Bemerkung 8.1). Der Kolben wird so lange erhitzt, bis alles Ammoniak überdestilliert ist.
5.2.2 Destillation von Borsäure
Wird der Ammoniakgehalt des Destillats von Hand titriert, findet das im Folgenden beschriebene Verfahren Anwendung. Bei einem voll automatisierten Destilliergerät, das auch die Titration des Ammoniakgehalts des Destillats vornimmt, ist die Betriebsanleitung des Herstellers zu befolgen.
Ein Auffangkolben mit 25 bis 30 ml Borsäurelösung ( 3.18) wird so unter den Ablauf des Kühlers gestellt, dass sich das Ablaufrohr unter der Oberfläche der überschüssigen Borsäurelösung befindet. Das Destilliergerät wird so eingestellt, dass es 50 ml Natriumhydroxidlösung ( 3.9) abgibt. Die Bedienung des Destilliergeräts erfolgt entsprechend den Anleitungen des Herstellers. Anschließend wird das durch die Zugabe der Natriumhydroxidlösung freigesetzte Ammoniak abdestilliert. Das Destillat wird in der Borsäure (Auffangsäure) aufgefangen. Die Destillatmenge (Dauer der Dampfdestillation) hängt von der in der Probe enthaltenen Menge Stickstoff ab. Hierbei sind die Anleitungen des Herstellers zu befolgen.
Anmerkung:
In einem halbautomatischen Destilliergerät erfolgen die Zugabe von überschüssigem Natriumhydroxid und die Dampfdestillation automatisch.
5.3 Titration
Es wird entweder gemäß 5.3.1 oder 5.3.2 weiterverfahren.
5.3.1 Schwefelsäure
Die überschüssige Schwefelsäure im Auffangkolben wird mit Natriumhydroxidlösung ( 3.10 oder 3.11) in Abhängigkeit von der Konzentration der verwendeten Schwefelsäure) titriert, bis der Endpunkt erreicht ist.
5.3.2 Borsäure
Der Inhalt des Auffangkolbens wird mit der volumetrischen Standardlösung der Salzsäure ( 3.19) oder mit der volumetrischen Standardlösung der Schwefelsäure ( 3.6) unter Verwendung einer Bürette titriert und die Menge des eingesetzten Titrationsmittels abgelesen.
Bei der kolorimetrischen Endpunktbestimmung ist der Endpunkt erreicht, wenn sich der Inhalt erstmals pink verfärbt. Die Bürette ist auf 0,05 ml genau abzulesen. Eine beleuchtete Magnetrührplatte oder ein fotometrischer Detektor können bei der Visualisierung des Endpunkts hilfreich sein.
Dies kann automatisch erfolgen bei Verwendung eines Wasserdampfdestillators mit automatischer Titration.
Hinsichtlich des Betriebs des jeweiligen Destillators bzw. des Destillier-/Titriergeräts sind die Anleitungen des Herstellers zu befolgen.
Anmerkung:
Bei Verwendung eines automatischen Titrationssystems beginnt die Titration unmittelbar mit dem Start der Destillation, und es wird die 1 %ige Borsäurelösung ( 3.18) eingesetzt.Wird ein vollautomatisches Destilliergerät eingesetzt, kann die automatische Titration des Ammoniaks auch mittels Endpunktbestimmung unter Verwendung eines potenziometrischen pH-Systems durchgeführt werden.
In diesem Fall wird eine automatische Titriervorrichtung mit einem pH-Meter verwendet. Das pH-Meter ist nach dem üblichen Labor-pH-Kalibrierverfahren ordnungsgemäß im Bereich pH 4 bis ph 7 zu kalibrieren.
Der pH-Endpunkt der Titration ist bei einem pH-Wert von 4,6 erreicht, dem steilsten Punkt der Titrationskurve (Wendepunkt).
5.4 Blindversuch
Zur Bestätigung, dass die Reagenzien stickstofffrei sind, wird ein Blindversuch (Aufschluss, Destillation und Titration) mit 1 g Saccharose ( 3.14) anstelle der Probe durchgeführt.
6. Berechnung der Ergebnisse
Die Berechnungen werden gemäß 6.1 oder 6.2 durchgeführt.
6.1 Berechnung für die Titration nach 5.3.1
Der Rohproteingehalt, ausgedrückt als Massenanteil, wird nach folgender Formel berechnet:
wobei:
V0 = Volumen NaOH (3.10 oder 3.11), das im Blindversuch eingesetzt wurde, in ml,
V1 = Volumen NaOH (3.10 oder 3.11), das in der Titration der Probe eingesetzt wurde, in ml,
c = Konzentration des Natriumhydroxids ( 3.10 oder 3.11), in mol/l,
m = Probeneinwaage in g.
6.2 Berechnung für die Titration nach 5.3.2
6.2.1 Titration mit Salzsäure
Der Rohproteingehalt, ausgedrückt als Massenanteil, wird nach folgender Formel berechnet: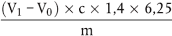
wobei:
m = Probeneinwaage in g,
c = Konzentration der volumetrischen Standardlösung von Salzsäure ( 3.19), in mol/l,
V0 = Volumen der Salzsäure, die im Blindversuch eingesetzt wurde, in ml,
V1 = Volumen der Salzsäure, die für die Probenmenge eingesetzt wurde, in ml.
6.2.2 Titration mit Schweflsäure
Der Rohproteingehalt, ausgedrückt als Massenanteil, wird nach folgender Formel berechnet: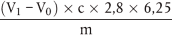
wobei:
m = Probeneinwaage in g,
c = Konzentration der volumetrischen Standardlösung von Schwefelsäure ( 3.6), in mol/l,
V0 = Volumen der Schwefelsäure ( 3.6), die im Blindversuch eingesetzt wurde, in ml,
V1 = Volumen der Schwefelsäure ( 3.6), die für die Probenmenge eingesetzt wurde, in ml.
7. Beurteilung des Verfahrens
7.1 Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
7.2 Genauigkeit
Die Analyse (Aufschluss, Destillation und Titration) wird mit 1,5 bis 2,0 g Acetanilid ( 3.13) in Gegenwart von 1 g Saccharose ( 3.14) durchgeführt; 1 g Acetanilid verbraucht 14,80 ml Schwefelsäure ( 3.5). Die Wiederfindung muss mindestens 99 % betragen.
8. Bemerkungen
8.1 Es können manuelle, halbautomatische oder automatische Geräte verwendet werden. Bei Geräten, die ein Umfüllen zwischen Aufschluss und Destillation erfordern, ist darauf zu achten, dass dies verlustlos geschieht. Verfügt der Destillationsapparat nicht über einen Tropftrichter, so erfolgt die Zugabe der Natriumhydroxidlösung unmittelbar vor dem Anschließen des Kolbens an den Kühler; die Flüssigkeit ist in diesem Fall langsam an den Kolbenwänden entlang laufen zu lassen.
8.2 Kommt es während des Aufschlusses zur Verfestigung der Mischung, ist mit der Bestimmung von vorn zu beginnen und eine größere als die oben genannte Menge an Schwefelsäure ( 3.4) zu verwenden.
8.3 Bei stickstoffarmen Proben kann die in den Auffangkolben einzufüllende Menge Schwefelsäure ( 3.7) gegebenenfalls auf 10 oder 15 ml verringert und mit Wasser auf 25 ml aufgefüllt werden.
8.4 Für Routineanalysen können auch andere Methoden zur Bestimmung des Rohproteins herangezogen werden; die in diesem Teil C beschriebene Kjeldahl-Methode ist jedoch die Referenzmethode. Die Gleichwertigkeit der Ergebnisse der alternativen Methode (z.B. DUMAS) im Vergleich zur Referenzmethode muss für jede Matrix einzeln nachgewiesen werden. Da die Ergebnisse einer alternativen Methode auch nach Feststellung ihrer Gleichwertigkeit leicht von den Ergebnissen der Referenzmethode abweichen können, muss im Analysebericht angegeben werden, welche Analyse zur Bestimmung des Rohproteins verwendet wurde.
D. Bestimmung des Harnstoffgehalts
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Harnstoffgehalts von Futtermitteln.
2. Prinzip
Die Probe wird unter Zusatz eines Klärungsmittels in Wasser suspendiert. Die Suspension wird filtriert. Nach Zugabe von 4-Dimethylaminobenzaldehyd (4-DMAB) wird der Gehalt an Harnstoff im Filtrat durch Messung der Extinktion bei einer Wellenlänge von 420 nm bestimmt.
3. Reagenzien
3.1 4-Dimethylaminobenzaldehydlösung: 1,6 g 4-DMAB werden in 100 ml 96 %igen Ethanols gelöst und 10 ml Salzsäure (ρ20 = 1,19 g/ml) hinzugefügt. Das Reagenz ist höchstens 2 Wochen haltbar.
3.2 Carrez-Lösung I: 21,9 g Zinkazetat, Zn(CH10COO)2.2H2O und 3 g Eisessig werden in Wasser gelöst und auf 100 ml mit Wasser aufgefüllt.
3.3 Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat (II), K4Fe(CN)6.3H2O, werden in Wasser gelöst und auf 100 ml mit Wasser aufgefüllt.
3.4 Aktivkohle, keinen Harnstoff adsorbierend (zu prüfen).
3.5 Harnstofflösung (Massenkonzentration = 0,1 %).
4. Geräte
4.1 Mechanisches Schüttelgerät, ca. 35 bis 40 min-1.
4.2 Reagenzgläser: 160 × 16 mm, mit Schliffstopfen.
4.3 Spektralfotometer.
5. Verfahren
5.1 Analysengang der Probe
Von der Probe werden 2 g auf 1 mg genau eingewogen und mit 1 g Aktivkohle ( 3.4) in einen 500-ml-Messkolben gebracht. Hierzu werden 400 ml Wasser und 5 ml Carrez-Lösung I ( 3.2) gegeben, ca. 30 s gemischt und dann 5 ml Carrez-Lösung II ( 3.3) zugesetzt. Das Ganze wird 30 min lang im Schüttelgerät rotiert. Dann wird mit Wasser zur Marke aufgefüllt, geschüttelt und filtriert.
Von den klaren und farblosen Filtraten werden je 5 ml in je ein Reagenzglas mit Schliffstopfen pipettiert, 5 ml 4-DMAB-Lösung (3.1) zugesetzt und gemischt. Die Gläser werden in einem Wasserbad bei 20 °C (+/4 °C) temperiert. Nach 15 min wird die Extinktion der Probenlösung im Vergleich mit der Blindprobenlösung der Reagenzien im Spektralfotometer bei 420 nm gemessen.
5.2 Kalibrationskurve
Volumen von 1, 2, 4, 5 bzw. 10 ml Harnstofflösung ( 3.5) werden entnommen, in je 100-ml-Messkolben gebracht und mit Wasser zur Marke aufgefüllt. Von jeder Lösung werden 5 ml mit je 5 ml 4-DMAB-Lösung (3.1) gemischt. Die Extinktion wird, wie oben angegeben, im Vergleich mit einer Lösung, die 5 ml 4-DMAB und 5 ml harnstofffreies Wasser enthält, gemessen. Es wird eine Kalibrationskurve aufgestellt.
6. Berechnung der Ergebnisse
Mithilfe der Kalibrationskurve ist die Menge an Harnstoff in der Versuchsprobe zu bestimmen.
Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
7. Bemerkungen
7.1 Bei einem Harnstoffgehalt von mehr als 3 % ist die Einwaage auf 1 g zu reduzieren bzw. die Anfangslösung so weit zu verdünnen, dass in 500 ml höchstens 50 mg Harnstoff enthalten sind.
7.2 Bei niedrigen Harnstoffgehalten wird die Einwaage erhöht, soweit das Filtrat klar und farblos bleibt.
7.3 Enthält die Probe einfache Stickstoffverbindungen, insbesondere Aminosäuren, so ist die Extinktion bei 435 nm zu messen.
E. Bestimmung des Gehalts an flüchtigen stickstoffhaltigen basen
I. Durch Mikrodiffusion
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an flüchtigen stickstoffhaltigen basen, ausgedrückt als Ammoniak, in Futtermitteln.
2. Prinzip
Die Probe wird mit Wasser extrahiert, die Lösung geklärt und filtriert. Die flüchtigen stickstoffhaltigen basen werden nach Zusatz von Kaliumcarbonatlösung durch Mikrodiffusion abgetrennt, in einer Borsäurelösung aufgefangen und mit Schwefelsäure titriert.
3. Reagenzien
3.1 Trichloressigsäurelösung (Massenkonzentration = 20 %).
3.2 Indikator: 33 mg Bromkresolgrün und 65 mg Methylrot werden in 100 ml Ethanol (Volumenkonzentration = 95 bis 96 %) gelöst.
3.3 Borsäurelösung: 10 g Borsäure werden in einem 1-l-Messkolben in 200 ml Ethanol (Volumenkonzentration = 95 bis 96 %) und 700 ml Wasser gelöst. Vom Indikator ( 3.2) werden 10 ml hinzugefügt. Die Lösung wird gemischt und nötigenfalls unter Zusatz von Natriumhydroxidlösung auf eine hellrote Farbe gebracht. Von dieser Lösung kann 1 ml bis zu 300 μg NH3 binden.
3.4 Gesättigte Kaliumkarbonatlösung: 100 g Kaliumcarbonat werden in 100 ml siedendem Wasser gelöst. Nach dem Abkühlen wird die Lösung filtriert.
3.5 0,01 mol/l Schwefelsäure.
4. Geräte
4.1 Mechanisches Schüttelgerät, ca. 35 bis 40 min-1.
4.2 Conwayschalen (vgl. Skizze) aus Glas oder Plastik.
4.3 Mikrobüretten mit 0,01-ml-Einteilung.
5. Verfahren
Von der Probe werden 10 g auf 1 mg genau gewogen, mit 100 ml Wasser in einen 200-ml-Messkolben gegeben und 30 min lang im Schüttelgerät gemischt oder verrührt. Dann werden 50 ml Trichloressigsäurelösung ( 3.1) hinzugefügt, mit Wasser zur Marke aufgefüllt, kräftig geschüttelt und durch einen Faltenfilter filtriert.
In die Mitte der Conwayschale wird 1 ml Borsäurelösung ( 3.3) und in den Ring der Schale 1 ml des Probenfiltrats pipettiert. Die Schale wird mit dem angefetteten Deckel teilweise bedeckt; dann wird schnell 1 ml der gesättigten Kaliumcarbonatlösung ( 3.4) in den Ring gegeben und die Schale luftdicht verschlossen. Um mit Sicherheit eine Mischung der beiden Reagenzien zu erreichen, wird die Schale vorsichtig in waagerechter Stellung gedreht. Darauf lässt man das Ganze mindestens 4 h lang bei Raumtemperatur oder 1 h lang bei 40 °C stehen.
Die in der Borsäurelösung enthaltenen flüchtigen basen werden anschließend mit Schwefelsäure ( 3.5) unter Verwendung einer Mikrobürette ( 4.3) titriert.
Nach demselben Verfahren ist ein Blindversuch ohne die zu analysierende Probe durchzuführen.
6. Berechnung der Ergebnisse
1 ml H2SO4 (0,01 mol/l) entspricht 0,34 mg Ammoniak.
Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
7. Bemerkung
Enthält die Probe mehr als 0,6 % Ammoniak, ist das ursprüngliche Filtrat zu verdünnen.
CONWAY CELL
Scale 1/1
II. Durch Destillation
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an flüchtigen stickstoffhaltigen basen, ausgedrückt als Ammoniak, in Fischmehl, das praktisch keinen Harnstoff enthält. Sie ist nur anzuwenden bei Gehalten < 0,25 % Ammoniak.
2. Prinzip
Die Probe wird mit Wasser extrahiert, die Lösung geklärt und filtriert. Die flüchtigen stickstoffhaltigen basen werden nach Zusatz von Magnesiumoxid in der Siedehitze abgetrennt und in einer definierten Menge Schwefelsäure aufgefangen, deren Überschuss durch Natriumhydroxidlösung zurücktitriert wird.
3. Reagenzien
3.1 Trichloressigsäurelösung (Massenkonzentration = 20 %).
3.2 Magnesiumoxid.
3.3 Antischaumemulsion (z.B. Silikon).
3.4 0,05 mol/l Schwefelsäure.
3.5 0,1 mol/l Natriumhydroxidlösung.
3.6 Methylrotlösung 0,3 %, in Ethanol (Volumenkonzentration = 95 bis 96 %).
4. Geräte
4.1 Mechanisches Schüttelgerät, ca. 35 bis 40 min-1.
4.2 Destillationsapparatur nach Kjeldahl.
5. Verfahren
Von der Probe werden 10 g auf 1 mg genau gewogen, mit 100 ml Wasser in einen 200-ml-Messkolben gegeben und 30 min lang im Schüttelgerät gemischt oder gerührt. Dann werden 50 ml Trichloressigsäurelösung ( 3.1) hinzugefügt, es wird mit Wasser zur Marke aufgefüllt, kräftig geschüttelt und durch einen Faltenfilter filtriert.
Je nach dem vermuteten Gehalt an flüchtigen stickstoffhaltigen basen wird eine entsprechende Menge (im Allgemeinen 100 ml) des klaren Filtrats auf 200 ml verdünnt und 2 g Magnesiumoxid ( 3.2) sowie einige Tropfen Antischaumemulsion ( 3.3) hinzugefügt. Die Lösung muss gegen Lackmuspapier alkalisch reagieren; anderenfalls ist noch Magnesiumoxid ( 3.2) zuzusetzen. Es ist gemäß 5.2 und 5.3 der Analysenmethode zur Bestimmung des Rohproteingehalts (Teil C dieses Anhangs) weiterzuverfahren.
Nach demselben Verfahren ist ein Blindversuch ohne die zu analysierende Probe durchzuführen.
6. Berechnung der Ergebnisse
1 ml H2SO4 (0,05 mol/l) entspricht 1,7 mg Ammoniak.
Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 10 % (relativ) Ammoniak nicht überschreiten.
F. Bestimmung des Gehalts an Aminosäuren (ausser Tryptophan)
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an freien (synthetischen und natürlichen) sowie der gesamten (peptidgebundenen und freien) Aminosäuren in Futtermitteln unter Verwendung eines Aminosäureanalysators.
Die Methode ist für folgende Aminosäuren anwendbar: Cyst(e)in, Methionin, Lysin, Threonin, Alanin, Arginin, Asparaginsäure, Glutaminsäure, Glycin, Histidin, Isoleucin, Leucin, Phenylalanin, Prolin, Serin, Tyrosin und Valin.
Die Methode unterscheidet nicht zwischen Salzen von Aminosäuren und dient auch nicht zur Unterscheidung der D- und L-Formen der Aminosäuren. Sie ist nicht zur Bestimmung des Gehalts an Tryptophan oder Hydroxyanaloga der Aminosäuren anwendbar.
2. Prinzip
2.1 Freie Aminosäuren
Die freien Aminosäuren werden mit verdünnter Salzsäure extrahiert. Mitextrahierte stickstoffhaltige Makromoleküle werden mit Sulfosalicylsäure ausgefällt und durch Filtrieren entfernt. Die filtrierte Lösung wird auf einen pH-Wert von 2,20 eingestellt. Die Aminosäuren werden durch Ionenaustauschchromatografie getrennt und nach Reaktion mit Ninhydrin durch fotometrischen Nachweis bei 570 nm bestimmt.
2.2 Gesamtaminosäuren
Die gewählte Methode hängt von den zu untersuchenden Aminosäuren ab. Cyst(e)in und Methionin müssen vor der Hydrolyse zu Cysteinsäure bzw. Methioninsulfon oxidiert werden. Tyrosin muss im Hydrolysat der nicht oxidierten Probe bestimmt werden. Alle übrigen unter 1 genannten Aminosäuren können aus dem Hydrolysat der oxidierten oder der nicht oxidierten Probe bestimmt werden.
Die Oxidation wird bei 0 °C mit einer Perameisensäure-Phenol-Mischung durchgeführt. Überschüssiges Oxidationsreagenz wird mit Natriumdisulfit zerstört. Die oxidierte bzw. die nicht oxidierte Probe wird mit Salzsäure ( 3.20) 23 h lang hydrolysiert. Das Hydrolysat wird auf einen pH-Wert von 2,20 eingestellt. Die Aminosäuren werden durch Ionenaustauschchromatografie getrennt und nach Reaktion mit Ninhydrin fotometrisch bei 570 nm (bei Prolin 440 nm) bestimmt.
3. Reagenzien
Es ist bidestilliertes Wasser oder Wasser gleichwertiger Qualität zu verwenden (Leitfähigkeit < 10 µS/cm).
3.1 Wasserstoffperoxid, w (Massenanteil) = 30 %.
3.2 Ameisensäure, w (Massenanteil) = 98 bis 100 %.
3.3 Phenol.
3.4 Natriumdisulfit.
3.5 Natriumhydroxid.
3.6 5-Sulfosalicylsäure-Dihydrat.
3.7 Salzsäure, Dichte ca. 1,18 g/ml.
3.8 Tri-Natriumcitrat-Dihydrat.
3.9 2,2' -Thiodiethanol (Thiodiglycol).
3.10 Natriumchlorid.
3.11 Ninhydrin.
3.12 Petrolether, Siedeintervall 40 bis 60 °C.
3.13 Norleucin oder sonstige für die Verwendung als interner Standard geeignete Verbindung.
3.14 Stickstoffgas (< 10 ppm Sauerstoff).
3.15 1-Octanol.
3.16 Aminosäuren.
3.16.1 Standardstoffe gemäß Auflistung unter 1. Es dürfen nur reine Verbindungen ohne Kristallwasser verwendet werden. Sie sind vor der Verwendung im Vakuum über P2O5 oder H2SO4 1 Woche lang zu trocknen.
3.16.2 Cysteinsäure.
3.16.3 Methioninsulfon.
3.17 Natriumhydroxidlösung, c = 7,5 mol/l:
300 g NaOH ( 3.5) in Wasser lösen und auf 1 l auffüllen.
3.18 Natriumhydroxidlösung, c = 1 mol/l:
40 g NaOH ( 3.5) in Wasser lösen und auf 1 l auffüllen.
3.19 Phenolhaltige Ameisensäurelösung:
889 g Ameisensäure ( 3.2) werden mit 111 g Wasser gemischt; anschließend werden 4,73 g Phenol ( 3.3) hinzugefügt.
3.20 Hydrolysemischung, c = 6 mol HCl/l unter Zusatz von 1 g Phenol/l:
Zu 492 ml HCl ( 3.7) wird 1 g Phenol ( 3.3) hinzugefügt und mit Wasser auf 1 l aufgefüllt.
3.21 Extraktionslösung, c = 0,1 mol HCl/l, 2 % Thiodiglycol enthaltend: 8,2 ml HCl ( 3.7) werden in ca. 900 ml Wasser gegeben, 20 ml Thiodiglycol ( 3.9) hinzugefügt, und es wird mit Wasser auf 1 l aufgefüllt ( 3.7 und 3.9 dürfen nicht unmittelbar gemischt werden).
3.22 5-Sulfosalicylsäurelösung, β = 6 %:
60 g 5-Sulfosalicylsäure ( 3.6) in Wasser lösen und mit Wasser auf 1 l auffüllen.
3.23 Oxidationsmischung (Perameisensäure-Phenol):
0,5 ml Wasserstoffperoxid ( 3.1) werden mit 4,5 ml phenolhaltiger Ameisensäurelösung ( 3.19) in einem kleinen Becherglas gemischt. Es wird bei 20 bis 30 °C 1 h stehen gelassen, um die Bildung von Perameisensäure zu bewirken. Anschließend wird die Lösung 15 min in ein Eisbad gestellt, bevor sie der Probe zugegeben wird.
Vorsicht: Hautkontakt vermeiden und Schutzkleidung tragen.
3.24 Citratpufferlösung, c = 0,2 mol Na+/l, pH 2,20:
19,61 g Natriumcitrat ( 3.8), 5 ml Thiodiglycol ( 3.9), 1 g Phenol (3.3) und 16,50 ml HCl ( 3.7) werden in ca. 800 ml Wasser gelöst. Der pH-Wert wird auf 2,20 eingestellt. Mit Wasser auf 1 l auffüllen.
3.25 Elutionspuffer entsprechend den Anforderungen des verwendeten Gerätes ( 4.9).
3.26 Ninhydrinreagenz entsprechend den Bedingungen des verwendeten Gerätes ( 4.9).
3.27 Aminosäuren-Standardlösungen: Diese Lösungen sind bei < 5 °C aufzubewahren.
3.27.1 Aminosäuren-Standard-Stammlösung ( 3.16.1):
c = 2,5 μmol/ml je Aminosäure in Salzsäure
Diese Lösung ist im Handel erhältlich.
3.27.2 Standard-Stammlösung von Cysteinsäure und Methioninsulfon, c = 1,25 μmol/ml:
0,2115 g Cysteinsäure ( 3.16.2) und 0,2265 g Methioninsulfon ( 3.16.3) werden in Citratpufferlösung ( 3.24) in einem 1.000-ml-Messkolben gelöst, und es wird mit Citratpufferlösung zur Marke aufgefüllt. Die Lösung ist bei < 5 °C höchstens 12 Monate haltbar. Sie wird nicht benötigt, falls die Standard-Stammlösung ( 3.27.1) bereits Cysteinsäure und Methioninsulfon enthält.
3.27.3 Stammlösung des internen Standards, z.B. Norleucin, c = 20 μmol/ml:
0,6560 g Norleucin ( 3.13) werden in Citratpufferlösung ( 3.24) in einem Messkolben gelöst, und es wird mit Citratpufferlösung auf 250 ml aufgefüllt. Diese Lösung ist bei < 5 °C höchstens 6 Monate haltbar.
3.27.4 Aminosäurenkalibrierlösung zur Verwendung bei Hydrolysaten, c = 5 nmol/50 μl Cysteinsäure und Methioninsulfon und c = 10 nmol/50 µl der übrigen Aminosäuren: 2,2 g Natriumchlorid (3.10) werden in einem 100-ml-Becherglas in 30 ml Citratpufferlösung ( 3.24) gelöst. Es werden 4,00 ml Standard-Stammlösung der Aminosäuren ( 3.27.1), 4,00 ml Standard-Stammlösung von Cysteinsäure und Methinoninsulfon ( 3.27.2), und, falls verwendet, 0,50 ml Stammlösung des internen Standards ( 3.27.3) hinzugefügt. Der pH-Wert wird mit Natriumhydroxid ( 3.18) auf 2,20 eingestellt.
Die Lösung wird quantitativ in einen 50-ml-Messkolben überführt, und es wird mit Citratpufferlösung ( 3.24) zur Marke aufgefüllt und gemischt.
Die Lösung ist bei < 5 °C höchstens 3 Monate haltbar.
Siehe auch Bemerkungen unter 9.1.
3.27.5 Aminosäurenkalibrierlösung zur Verwendung bei Hydrolysaten gemäß 5.3.3.1 und bei Extrakten gemäß 5.2. Die Kalibrierlösung wird gemäß 3.27.4 hergestellt, jedoch ohne Zugabe von Natriumchlorid.
Die Lösung ist bei < 5 °C höchstens 3 Monate haltbar.
4. Geräte
4.1 100- oder 250-ml-Rundkolben mit Rückflusskühler.
4.2 100-ml-Borosilikat-Glasflaschen mit Schraubverschluss mit Gummidichtung/teflonbeschichteter Dichtung (z.B. Duran, Schott).
4.3 Trockenschrank mit forcierter Umluft, auf ± 2 °C regelbar.
4.4 pH-Meter (auf drei Dezimalstellen genau).
4.5 Membranfilter, 0,22m Porengröße.
4.6 Zentrifuge.
4.7 Vakuum-Rotationsverdampfer.
4.8 Mechanischer Schüttler oder Magnetrührer.
4.9 Aminosäureanalysator oder HPLC-Einrichtung mit Ionenaustauschersäule, Einrichtung für Ninhydrin-Nachsäulenderivatisierung und Fotometer.
Die Säule wird mit sulfoniertem Polystyrolharz gefüllt, das die Trennung der einzelnen Aminosäuren und deren Abtrennung von sonstigen Ninhydrinpositiven Substanzen ermöglicht. Die Pumpen für die Ninhydrin- und für die Pufferlösungen müssen über den Zeitraum des Kalibrationslaufes und des Probenlaufes eine Flussstabilität von ± 0,5 % gewährleisten.
Bei einigen Aminosäureanalysatoren können auch Hydrolyseverfahren angewandt werden, wenn die Hydrolysate Natriumionenkonzentrationen von c = 0,8 mol/l aufweisen und die restliche Ameisensäure des Oxidationsschrittes enthalten. Andere Analysatoren bieten keine befriedigende Abtrennung bestimmter Aminosäuren, falls das Hydrolysat überschüssige Ameisensäure und/oder hohe Natriumionenkonzentrationen aufweist. In diesem Fall wird das Säurevolumen nach der Hydrolyse und vor der pH-Wert-Einstellung durch Einengen auf ca. 5 ml reduziert. Das Einengen ist unter Vakuum bei höchstens 40 °C vorzunehmen.
 |
weiter . |  |
(Stand: 29.10.2020)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: 90.- € netto (Grundlizenz)
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion