 Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 1996, Lebensmittel - EU Bund

| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 1996, Lebensmittel - EU Bund |
|
Richtlinie 96/73/EG des Europäischen Parlaments und des Rates vom 16. Dezember 1996 über bestimmte Methoden der quantitativen Analyse von binären Textilfasergemischen
(ABl. Nr. L 32 vom 03.02.1997 S. 1;
VO (EG) 1882/2003 - ABl. Nr. L 284 vom 13.10.2003 S. 1;
RL 2006/2/EG - ABl. Nr. L 5 vom 10.01.2006 S. 10;
RL 2007/4/EG - ABl. Nr. L 28 vom 03.02.2007 S. 14;
VO (EG) 1137/2008 - ABl. Nr. L 5 vom 10.01.2006 S. 1;
RL 2009/122/EG - ABl. Nr. L 5 vom 10.01.2006 S. 14;
RL 2011/74/EU - ABl. Nr. L 198 vom 30.07.2011 S. 32;
VO (EU) 1007/2011 - ABl. Nr. L 272 vom 18.10.2011 S. 1 Inkrafttreten Gültig Übergangsbestimmung Entsprechungstabellen aufgehoben)
aufgehoben/ersetzt gemäß Art. 27 durch VO (EU) 1007/2011 - (ABl. Nr. L 272 vom 18.10.2011 S. 1) Inkrafttreten/Gültig Übergangsbestimmung Entsprechungstabellen
Das Europäische Parlament und der Rat der Europäischen Union -
gestützt auf den Vertrag zur Gründung der Europäischen Gemeinschaft, insbesondere auf Artikel 100a,
auf Vorschlag der Kommission 1,
nach Stellungnahme des Wirtschafts- und Sozialausschusses 2, gemäß dem Verfahren des Artikels 189b des Vertrags 3,
in Erwägung nachstehender Gründe:
Die Richtlinie 72/276/EWG des Rates vom 17. Juli 1972 zur Angleichung der Rechtsvorschriften der Mitgliedstaaten über bestimmte Methoden der quantitativen Analyse von binären Textilfasergemischen 4 ist mehrfach in wesentlichen Punkten geändert worden. Aus Gründen der Klarheit und der Übersichtlichkeit empfiehlt es sich daher, sie zu kodifizieren.
Die Richtlinie 96/74/EG des Europäischen Parlaments und des Rates vom 16. Dezember 1996 zur Bezeichnung von Textilerzeugnissen 5 sieht eine Kennzeichnungspflicht für die Faserzusammensetzung von Textilerzeugnissen vor; die Übereinstimmung dieser Erzeugnisse mit den Angaben auf dem Etikett wird mittels Analyse geprüft.
Bei amtlichen Kontrollen in den Mitgliedstaaten müssen einheitliche Methoden zur Bestimmung der Faserzusammensetzung der Textilerzeugnisse angewandt werden, und zwar sowohl in bezug auf die Vorbehandlung der Probe als auch bei der quantitativen Analyse.
Die Richtlinie 96/74/EG sieht vor, daß in besonderen Richtlinien die Methoden der Probenahme und die Analysemethoden angegeben werden, die in allen Mitgliedstaaten zur Bestimmung der Faserzusammensetzung der Erzeugnisse anzuwenden sind. Infolgedessen werden in Anhang II der vorliegenden Richtlinie fünfzehn einheitliche Analysemethoden für die meisten auf dem Markt vorhandenen Textilerzeugnisse aus binären Gemischen festgelegt.
Der technische Fortschritt macht eine häufige Anpassung der in den Einzelrichtlinien über die Analysemethoden auf dem Textilsektor aufgeführten technischen Vorschriften erforderlich. Um die Durchführung der hierfür erforderlichen Maßnahmen zu erleichtern, muß ein Verfahren geschaffen werden, das eine enge Zusammenarbeit zwischen Mitgliedstaaten und Kommission im Rahmen des Ausschusses für den Bereich der Richtlinien über die Bezeichnung und Etikettierung von Textilerzeugnissen vorsieht.
Bei binären Gemischen, für die keine einheitliche Analysemethode auf Gemeinschaftsebene besteht, wird die Zusammensetzung dieser Gemische von dem mit der Prüfung betrauten Laboratorium mit Hilfe einer ihm zur Verfügung stehenden geeigneten Methode bestimmt, wobei in dem Analysebericht das erzielte Ergebnis und die bei der Methode gegebene Genauigkeit, soweit sie bekannt ist, angegeben werden.
Die Bestimmungen dieser Richtlinie stimmen mit der Stellungnahme des Ausschusses für den Bereich der Richtlinien über die Bezeichnung und Etikettierung von Textilerzeugnissen überein.
Diese Richtlinie darf nicht die Pflichten der Mitgliedstaaten hinsichtlich der in Teil B des Anhangs III genannten Umsetzungsfristen berühren
- haben folgende Richtlinie erlassen:
Diese Richtlinie betrifft Methoden zur quantitativen Analyse von bestimmten binären Textilfasergemischen und die Vorbereitung von Vorproben und Analyseproben.
Unter Vorprobe ist eine für Analysezwecke geeignete Teilprobe aus den Laboratoriumssammelproben zu verstehen, die ihrerseits aus einer Lieferung des zu prüfenden Gutes entnommen worden ist.
Die Analyseprobe ist der Teil der Vorprobe, der zur Erzielung eines Analyseergebnisses im Einzelfall erforderlich ist.
Die Mitgliedstaaten treffen gemäß der Richtlinie 96/74/EG alle erforderlichen Maßnahmen, damit bei amtlichen Prüfungen zur Bestimmung der Zusammensetzung von auf den Markt gebrachten Textilerzeugnissen die Bestimmungen der Anhänge I und II über die quantitativen Analysemethoden für bestimmte binäre Textilfasergemische einschließlich der Vorbereitung der Vorproben und Analyseproben befolgt werden.
Das Laboratorium, das mit der Prüfung eines binären Gemisches, für das keine einheitliche Analysemethode auf Gemeinschaftsebene besteht, betraut ist, bestimmt die Zusammensetzung dieses Gemisches mit Hilfe einer ihm zur Verfügung stehenden geeigneten Methode und gibt in dem Analysebericht das erzielte Ergebnis und die bei der Methode gegebene Genauigkeit, soweit sie bekannt ist, an.
Die Kommission passt die in Anhang II vorgesehenen quantitativen Analysemethoden an den technischen Fortschritt an. Diese Maßnahmen zur Änderung nicht wesentlicher Bestimmungen dieser Richtlinie werden nach dem in Artikel 6 Absatz 2 genannten Regelungsverfahren mit Kontrolle erlassen.
(1) Die Kommission wird von einem Ausschuss für den Bereich der Richtlinien über die Bezeichnung und Etikettierung von Textilerzeugnissen unterstützt.
(2) Wird auf diesen Absatz Bezug genommen, so gelten Artikel 5a Absätze 1 bis 4 und Artikel 7 des Beschlusses 1999/468/EG unter Beachtung von dessen Artikel 8.
Die Mitgliedstaaten teilen der Kommission den Wortlaut der wichtigsten innerstaatlichen Rechtsvorschriften mit, die sie auf dem unter diese Richtlinie fallenden Gebiet erlassen.
Die in Teil A des Anhangs III aufgeführten Richtlinien werden unbeschadet der Pflichten der Mitgliedstaaten hinsichtlich der in Teil B des Anhangs III genannten Umsetzungsfristen aufgehoben.
Bezugnahmen auf die aufgehobenen Richtlinien gelten als Bezugnahmen auf diese Richtlinie und sind nach Maßgabe der Übereinstimmungstabelle im Anhang IV zu lesen.
Diese Richtlinie ist an die Mitgliedstaaten gerichtet.
Sie tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Gemeinschaften in Kraft.
2) ABl. Nr. C 195 vom 18.07.1994 S. 10.
3) Stellungnahme des Europäischen Parlaments vom 15. Februar 1995 (ABl. Nr. C 56 vom 06.03.1995 S. 53), gemeinsamer Standpunkt des Rates vom 26. Februar 1996 (ABl. Nr. C 196 vom 06.07.1996 S. 20), Beschluß des Europäischen Parlaments vom 18. Juni 1996 (ABl. Nr. C 198 vom 08.07.1996 S. 25), Beschluß des Rates vom 7. Oktober 1996.
4) ABl. Nr. L 173 vom 31.07.1972 S. 1. Richtlinie zuletzt geändert durch die Richtlinie 87/184/EWG (ABl. Nr. L 75 vom 17.03.1987 S. 21).
5) Siehe Seite 38 dieses Amtsblatts.
| Vorbereitung der Vorproben und der Analysenproben zur Bestimmung der Zusammensetzung von Textilerzeugnissen | Anhang I |
1. Anwendungsbereich
Dieser Anhang enthält allgemeine Hinweise für die Herstellung von Vorproben geeigneter Größe (d. h. nicht über 100 g) zur Vorbehandlung für quantitative Analysen aus Laboratoriumssammelproben sowie für die Auswahl von Analysenproben aus Vorproben, aus denen die nichtfaserigen Bestandteile in einer Vorbehandlung entfernt worden sind 1.
2. Begriffsbestimmungen
2.1 Prüfgut - Diejenige Materialmenge, die aufgrund einer Reihe von Untersuchungsergebnissen beurteilt werden soll. Dazu kann zum Beispiel das gesamte Material einer Stofflieferung gehören, das gesamte von einer bestimmten Maschine gewebte Gewebe, eine Sendung Garne oder ein Ballen bzw. eine aus mehreren Ballen bestehende Lieferung.
2.2 Laboratoriumssammelprobe - Teil des Prüfguts, der als repräsentativ aus der Gesamtmenge entnommen worden ist und an das Laboratorium eingesandt wird. Größe und Art der Laboratoriumssammelprobe sind so zu wählen, daß sie die Schwankungen innerhalb der Lieferung richtig wiedergibt und im Laboratorium leicht zu handhaben ist 2.
2.3 Vorprobe - Teil der Laboratoriumssammelprobe, aus dem in einer Vorbehandlung die nichtfaserigen Bestandteile entfernt und anschließend die Analysenproben entnommen werden 3. Größe und Art der Vorprobe sind so zu wählen, daß sie die Schwankungen innerhalb der Laboratoriumssammelprobe richtig wiedergibt.
2.4 Analysenprobe - Teil der Vorprobe, der für die quantitative Einzelanalyse erforderlich ist.
3. Prinzip
Die Vorprobe wird so ausgewählt, daß sie die Laboratoriumssammelprobe repräsentiert.
Die Analysenproben werden aus der Vorprobe so ausgewählt, daß sie die letztere repräsentieren.
4. Probenahme aus losen Fasern
4.1 Ungerichtete Fasern - Die Vorprobe wird aus zufallsbedingt der Laboratoriumssammelprobe entnommenen Büscheln zusammengestellt. Die gesamte Vorprobe mischt man gründlich mit Hilfe einer Laboratoriumskrempel 4. Die Krempelflor sowie die noch in der Krempel hängenden Fasern und der aus der Krempel herausgefallene Faserbruch werden vorbehandelt. Anschließend werden die Analysenproben im jeweiligen Gewichtsverhältnis aus dem Krempelflor, den anhängenden Fasern und den herausgefallenen Fasern entnommen.
Bleibt die Form des Krempelflors durch die Vorbehandlung im wesentlichen unverändert, so werden die Analysenproben in der unter Punkt 4.2 beschriebenen Weise entnommen. Wird der Krempelflor durch die Vorbehandlung zerstört, so werden für die Restproben mindestens 16 kleine Büschel von geeigneter und unter sich möglichst gleicher Größe aus der vorbehandelten Probe herausgenommen und zu einer Probe zusammengefaßt.
4.2 Gerichtete Fasern (Krempelflor, Kammzug, Vorgarne) - Aus zufällig ausgewählten Teilen der Laboratoriumssammelprobe werden mindestens 10 Schnittproben zu je etwa 1 g hergestellt. Die so entstandene Vorprobe wird vorbehandelt. Dann werden die Schnittproben Kante an Kante aneinandergelegt und daraus die Analysenproben in der Weise hergestellt, daß jeweils ein Schnitt so durch die 10 Muster gelegt wird, daß von jeder der 10 Längen ein Teil erfaßt wird.
5. Probenahme aus Garnen
5.1 Garne auf Hülsen oder in Strängen - Es müssen alle Hülsen oder Stränge der Laboratoriumssammelprobe verwendet werden.
Man entnimmt jeder Hülse oder jedem Strang entsprechend zusammenhängende Fäden gleicher Länge, indem man Stränge gleicher Zahl auf eine Haspel 5 aufwindet oder auf andere Weise. Die einzelnen Längen werden zu einem einzigen Strang oder Kabel zusammengelegt, wobei darauf zu achten ist, daß in jedem Strang oder Kabel immer gleiche Fadenlängen von jeder Hülse oder jedem Strang vorhanden sind.
Die auf diese Weise erhaltene Vorprobe wird vorbehandelt.
Zur Entnahme von Analysenproben aus der vorbehandelten Vorprobe werden Fadenabschnitte gleicher Menge aus dem Strang oder Kabel herausgenommen; dabei ist darauf zu achten, daß keiner der darin enthaltenen Fäden ausgelassen wird.
Istt die Feinheit in "Tex" undn die Anzahl der Hülsen oder Stränge der Laboratoriumssammelprobe, so beträgt die Fadenlänge von jeder Hülse oder jedem Strang, die eine Vorprobe von 10 g ergibt,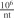 cm.
cm.
Ist nt sehr hoch, zum Beispiel über 2000, so wird ein schwerer Fadenstrang aufgewunden und in zwei Teile zerschnitten, um einen Strang von geeignetem Gewicht herzustellen. Die Enden einer Probe in Strangform sind vor Beginn der Vorbehandlung sorgfältig zusammenzubinden; die Analysenproben sind an einer Stelle zu entnehmen, die von dem abgebundenen Ende genügend weit entfernt ist.
5.2 Kettfäden - Die Vorprobe wird in der Weise entnommen, daß man vom Ende der Kette ein Stück abschneidet, das mindestens 20 cm lang ist und alle Kettfäden mit Ausnahme der Webkante enthält, die verworfen wird. Man bündelt einige Fäden an einem Ende zusammen. Ist die Probe zu schwer, um im ganzen vorbehandelt zu werden, so wird sie in zwei oder mehr Teile unterteilt, wobei jedes Teil vor der Vorbehandlung zusammengebunden wird. Die einzelnen Teile werden getrennt vorbehandelt und danach wieder zusammengefaßt. Von der Vorprobe wird eine Analysenprobe von passender Länge in genügendem Abstand von der Bündelung abgeschnitten, wobei darauf zu achten ist, daß keiner der Kettfäden ausgelassen wird. Bei einer Kette mitn Fäden der Feinheitt in "Tex" beträgt die Länge einer Probe von 1 g Gewicht cm.
cm.
6. Probenahme aus textilen Flächengebilden
6.1 Laboratoriumssammelprobe bestehend aus einem einzigen repräsentativen Abschnitt - Man schneidet einen diagonalen Streifen von Ecke zu Ecke und entfernt die Webkanten. Dieser Streifen ist die Vorprobe. Für eine Vorprobe x g beträgt die Fläche des Streifens cm.
cm.
m = Flächengewicht des Gewebes in g/m2.
Nach der Vorbehandlung zerschneidet man den Streifen quer zur Länge in 4 gleiche Teile und legt diese übereinander.
Man entnimmt Analysenproben aus einem beliebigen Teil des übereinandergeschichteten Materials, in dem man alle Lagen in der Weise durchschneidet, daß man von jeder Lage eine Analysenprobe mit gleicher Länge erhält.
Enthält der Stoff ein gewebtes Muster, so darf die Breite der Vorprobe, parallel zur Richtung der Kette gemessen, nicht kleiner sein als eine Wiederholung des Musters in der Kette. Ist unter diesen Bedingungen die Vorprobe zu groß, um im ganzen vorbehandelt zu werden, so muß sie in gleiche Teile zerschnitten werden, die getrennt vorzubehandeln sind; diese Teile sind vor der Herstellung der Analysenprobe übereinanderzulegen, doch ist darauf zu achten, daß entsprechende Teile des Musters nicht zusammenfallen.
6.2 Laboratoriumssammelproben, die aus mehreren Stoffabschnitten bestehen - Jeder Abschnitt wird gemäß 6.1 analysiert; die Ergebnisse werden getrennt angegeben.
7. Probenahme von Enderzeugnissen und/oder Konfektionsartikeln
Die Laboratoriumssammelprobe besteht in der Regel aus einem ganzen Konfektionsartikel oder aus einem repräsentativen Teilstück des Artikels.
Gegebenenfalls ist der Prozentsatz der verschiedenen Teile zu bestimmen, die nicht den gleichen Fasergehalt haben, damit festgestellt werden kann, ob Artikel 9 der Richtlinie 96/74/EG des Europäischen Parlaments und des Rates vom 16. Dezember 1996 zur Bezeichnung von Textilerzeugnissen anwendbar ist.
Dem Teil des Enderzeugnisses bzw. des Konfektionsartikels, dessen Zusammensetzung durch ein Etikett gekennzeichnet werden soll, ist eine repräsentative Vorprobe zu entnehmen. Trägt der Fertigartikel mehrere Etiketten, so müssen repräsentative Vorproben aus jedem Teil, der durch ein Etikett bezeichnet werden soll, entnommen werden.
Ist der Artikel, dessen Zusammensetzung bestimmt werden soll, nicht homogen, so kann es erforderlich sein, aus jedem Teil des Artikels Vorproben zu entnehmen und den proportionalen Anteil der einzelnen Teile, bezogen auf den ganzen Artikel, zu bestimmen.
Die Prozentsätze werden dann unter Berücksichtigung der verhältnismäßigen Anteile der untersuchten Teile errechnet.
Diese Vorproben werden vorbehandelt.
Anschließend werden den vorbehandelten Vorproben repräsentative Analysenproben entnommen.
_________
1) Gegebenenfalls kann man direkt die Analysenproben vorbehandeln.
2) Für Enderzeugnisse und Konfektionsartikel siehe Punkt 7.
3) Siehe Punkt 1.
4) Statt mit einer Laboratoriumskrempel kann auch mit einem Fasermischer gearbeitet oder das Verfahren der "ausgekämmten Büschel" (Hecheln des Doublierens, Teilens und anteiliges Verwerfen) angewendet werden.
5) Bei Verwendung in einer geeigneten Haspel können mehrere Hülsen gleichzeitig aufgewunden werden.
| Quantitative Analysenmethoden für bestimmte binäre Textilfasergemische | Anhang II |
1. Allgemeiner Teil
Einleitung
Die quantitativen Analysenmethoden bei Textilfasermischungen stützen sich hauptsächlich auf zwei Verfahren, nämlich auf das der manuellen Trennung und das der chemischen Trennung der Fasern.
Dem manuellen Trennverfahren sollte nach Möglichkeit der Vorzug gegeben werden, denn im allgemeinen führt es zu genaueren Ergebnissen als das chemische Verfahren. Das manuelle Verfahren läßt sich auf alle Textilerzeugnisse, bei denen die dieses Erzeugnis bildenden Fasern keine untrennbare Mischung darstellen, anwenden, z.B. auf die aus mehreren Bestandteilen zusammengesetzten Garne, bei denen die einzelnen Bestandteile aus einer einzigen Faserart gebildet werden, sowie auf Gewebe, bei denen die Kette aus einer anderen Faser als der Schuß besteht, oder auf Gewirke, die aus verschiedenartigen Garnen zusammengesetzt sind.
In der Regel stützt sich die quantitative chemische Analysenmethode bei Textilfasergemischen auf die selektive Lösbarkeit der Einzelbestandteile der Mischung. Nach Entfernung eines Bestandteils wird der unlösliche Rückstand gewogen, und der Anteil des löslichen Bestandteils wird unter Zugrundelegung des Gewichtsverlusts berechnet. Im vorliegenden Dokument werden die Angaben, die sich generell bei einer Analyse nach diesem Verfahren ergeben und für die im vorliegenen Anhang genannten Fasergemische jeder Zusammensetzung gelten, zusammengestellt. Dieses Dokument muß daher in Verbindung mit den anderen Texten benutzt werden, die ausführliche Verfahren für besondere Fasermischungen enthalten. Es kann vorkommen, daß bestimmte chemische Analysen auf anderen Prinzipien als dem der selektiven Auflösbarkeit beruhen. In diesem Fall werden in dem entsprechenden Teil der einschlägigen Methode ausführliche Angaben hierüber gemacht.
Die während der Herstellung der Textilerzeugnisse benutzten Fasergemische und in geringerem Maße auch die Fasergemische in den fertigen Textilien können oftmals als natürliche Beimengungen oder Erleichterung des Verarbeitungsvorganges Fette, Wachse oder Hilfsstoffe bzw. wasserlösliche Stoffe enthalten. Diese nicht zur Faser gehörenden Stoffe müssen vor der Analyse ausgesondert werden. Aus diesem Grund wird ebenfalls eine Methode der Vorbehandlung zwecks Entfernung von Ölen, Fetten, Wachsen und wasserlöslichen Stoffen angegeben.
Textilien können außerdem Kunstharze oder andere Stoffe enthalten, die ihnen bestimmte Eigenschaften verleihen sollen. Diese Stoffe, zu denen in Ausnahmefällen auch Farbstoffe gehören, können die Einwirkung des Reagenzes auf die löslichen Bestandteile beeinträchtigen bzw. teilweise oder vollständig durch das Reagenz beseitigt werden. Diese Zusatzstoffe können somit Fehler hervorrufen und müssen vor der Analyse der Probe ausgeschieden werden. Ist dies unmöglich, so sind die in diesem Anhang beschriebenen Verfahren der quantitativen Analyse nicht mehr anwendbar.
Farbstoffe in gefärbten Fasern werden als integrierender Bestandteil der Faser angesehen und nicht entfernt.
Bei diesen Analysen wird vom Trockengewicht ausgegangen; es wird daher ein Verfahren zur Bestimmung des Trockengewichts angegeben.
Bei der Ergebnisdarstellung werden zum Trockengewicht der einzelnen Fasern die in Anhang II der Richtlinie 96/74/EG des Europäischen Parlamentes und des Rates vom 16. Dezember 1996 zur Bezeichnung von Textilerzeugnisssen angegebenen Feuchtigkeitszuschläge zugerechnet.
Die in der Mischung enthaltenen Fasern sind vor der Analyse zu identifizieren. Bei bestimmten chemischen Methoden kann der unlösliche Bestandteil von Gemischen teilweise in dem Reagenz aufgelöst werden, das zur Auflösung der löslichen Bestandteile verwendet wird. Nach Möglichkeit wurden die Reagenzien so gewählt, daß sie nur einen geringen oder überhaupt keinen Einfluß auf die unlöslichen Fasern haben. Ist bei der Analyse mit einem Gewichtsverlust zu rechnen, so müssen die Ergebnisse entsprechend korrigiert werden; Korrekturfaktoren hierfür sind angegeben. Diese Korrekturfaktoren wurden in mehreren Laboratorien dadurch bestimmt, daß durch Vorbehandlung gereinigte Fasern mit dem entsprechenden Reagenz unter Befolgung der Analysenmethode behandelt wurden.
Diese Korrekturfaktoren gelten nur für normale Fasern, und weitere Korrekturfaktoren können erforderlich sei, wenn die Fasern vor oder während der Verarbeitung nicht intakt geblieben sind. Die angegebenen chemischen Analysenmethoden gelten für Einzelanalysen. Es muß mindestens an zwei getrennten Analysenproben je eine Analyse sowohl beim manuellen Trennungsverfahren als auch beim chemischen Trennungsverfahren durchgeführt werden. In Zweifelsfällen muß, sofern dies nicht technisch unmöglich ist, eine weitere Analyse durchgeführt werden, und zwar nach einem Verfahren, mit dem sich die bei dem ersten Verfahren als Rückstand gebildete Faser auflösen läßt.
I. Allgemeines über die Quantitativ-Chemischen Analysenmethoden für Fasergemische
Generelle Angaben zu den Verfahren der quantitativen chemischen Analyse von Fasergemischen
I.1. Anwendungsbereich
Unter dem "Anwendungsbereich" jeder Methode werden die Fasern aufgeführt, auf die sie anzuwenden ist.
I.2. Prinzip
Nach Identifizierung der einzelnen Bestandteile der Fasergemische wird einer der beiden Bestandteile entfernt, und zwar in der Regel durch selektive Auflösung1, wobei der unlösliche Rückstand gewogen und der Anteil des löslichen Bestandteils aus dem Gewichtsverlust berechnet wird. Außer bei technischen Schwierigkeiten ist vorzugsweise die in größerer Menge vorhandene Faser aufzulösen, damit man die in geringerer Menge vorhandene Faser als Rückstand erhält.
I.3. Erforderliches Material
I.3.1.Geräte
I.3.1.1. Filtertiegel und Wägegläser zum Einsetzen von Tiegeln oder andere gleichwertige Geräte
I.3.1.2. Absaugflasche
I.3.1.3. Exsikkator mit gefärbtem Kieselgel als Feuchtigkeitsindikator
I.3.1.4 Trockenofen mit Ventilator zur Trocknung der Analysenproben bei (105 ± 3) °C
I.3.1.5. Analysenwaage, Empfindlichkeit 0,0002 g
I.3.1.6. Extraktionsapparat Soxhlet oder gleichwertige Apparatur
I.3.2.Reagenzien
I.3.2.1 Petroläther, nachdestillliert, Siedebereich 40 bis 60 °C
I.3.2.2. Sonstige Reagenzien sind in den entsprechenden Teilen der Methode angegeben. Alle Reagenzien müssen chemisch rein sein
I.3.2.3. Destilliertes oder entionisiertes Wasser
I.3.2.4. Aceton
I.3.2.5. Orthophosphorsäure
I.3.2.6. Harnstoff
I.3.2.7. Natriumbicarbonat
I.4. Konditionierungs- und Analysenatmosphäre
Da die Trockenmasse bestimmt wird, ist weder eine Konditionierung der Probe noch eine Untersuchung in klimatisierter Atmosphäre erforderlich.
I.5. Vorprobe
Es wird eine für die Laboratoriumsprobe repräsentative Vorprobe gewählt, die für sämtliche erforderlichen Analysenproben von jeweils mindestens 1 g ausreicht.
I.6. Vorbehandlung der Vorprobe2
Ist einer der bei der Berechnung der Prozentsätze nicht zu berücksichtigenden Bestandteile vorhanden (siehe Artikel 12 Unterabsatz 3 der Richtlinie 96/74/EG des Europäischen Parlaments und des Rates vom 16. Dezember 1996 zur Bezeichnung von Textilerzeugnissen), so ist dieser zunächst durch eine geeignete Methode zu entfernen, die jedoch keinen der Faserbestandteile angreifen darf.
Zu diesem Zweck werden die mit Hilfe von Petrolether und Wasser extrahierbaren nichtfaserigen Bestandteile entfernt, indem die luftgetrocknete Probe im Soxhlet-Apparat mit Petrolether während einer Stunde und mit mindestens sechs Umläufen pro Stunde behandelt wird. Anschließend wird der Petrolether der Probe verdampft; danach wird die Probe durch Direktbehandlung extrahiert, das heißt durch einstündiges Eintauchen in Wasser bei Zimmertemperatur mit darauf folgendem einstündigen Eintauchen in Wasser bei 65 ± 5 °C unter zeitweiligem Schütteln, Flottenverhältnis 1:100. Danach wird das überschüssige Wasser durch Ausquetschen, Absaugen oder Zentrifugieren entfernt, bis die Probe lufttrocken ist.
Bei Elastolefin oder Fasergemischen, die Elastolefin und andere Fasern enthalten (Wolle, Tierhaare, Seide, Baumwolle, Flachs, Hanf, Jute, Manila, Alfa, Kokos, Ginster, Ramie, Sisal, Cupro, Modal, regenerierte Proteinfasern, Viskose, Polyacryl, Polyamid oder Nylon, Polyester, Elastomultiester), sollte das oben beschriebene Verfahren dahingehend leicht abgeändert werden, dass Petrolether durch Aceton ersetzt wird.
Bei binären Mischungen, die Elastolefin und Acetat enthalten, ist folgende Vorbehandlung durchzuführen. Die Probe wird 10 Minuten lang bei 80 °C mit einer Lösung extrahiert, die 25 g/l 50 %ige Orthophosphorsäure und 50 g/l Harnstoff enthält, Flottenverhältnis 1:100. Die Probe wird in Wasser gewaschen, danach lässt man sie abtropfen und wäscht sie in einer 0,1 %igen Natriumbicarbonat-Lösung sowie abschließend vorsichtig in Wasser.
Falls die nichtfaserigen Bestandteile nicht mit Hilfe von Petrolether und Wasser extrahiert werden können, so müssen sie anstatt mit Wasser, wie oben beschrieben, mit einem geeigneten Stoff entfernt werden, der keinen der Faserbestandteile wesentlich verändert. Bei einigen natürlichen Pflanzen-Rohfasern (wie zum Beispiel Jute-, Kokosfasern) ist zu beachten, dass durch die normale Vorbehandlung mit Petrolether und Wasser nicht alle natürlichen nichtfaserigen Bestandteile ausgesondert werden. Trotzdem werden keine weiteren Vorbehandlungen vorgenommen, soweit die Probe keine in Petrolether und in Wasser unlöslichen Appreturen enthält.
In den Analysenberichten müssen die gewählten Vorbehandlungsmethoden eingehend geschildert werden.
I.7. Analysengang
I.7.1. Allgemeine Anweisungen
I.7.1.1. Trocknung
Alle Trockenoperationen sind mindestens 4 Stunden, jedoch nicht mehr als 16 Stunden, bei (105 ± 3) °C in einem belüfteten Ofen bei geschlossener Ofentür durchzuführen. Beträgt die Trocknungsdauer weniger als 14 Stunden, muß überprüft werden, ob ein konstantes Gewicht erreicht wurde. Dieses Gewicht kann als erreicht gelten, wenn der Gewichtsunterschied nach einer neuen Trocknung von 60 Minuten weniger als 0,05% beträgt.
Die Filtertiegel und Wägegläser sowie die Proben oder die Rückstände sollen während des Trocknungs-, Abkühlungs- und Wägevorgangs nicht mit bloßen Händen berührt werden.
Die Analysenproben werden in einem Wägeglas mit abgenommenen Stopfen getrocknet. Nach der Trocknung wird das Wägeglas vor Herausnahme aus dem Ofen geschlossen und so schnell wie möglich in den Exsikkator gebracht.
Der Filtertiegel, der mit seinem Deckel in einem Wägeglas untergebracht ist, wird im Ofen getrocknet. Nach der Trocknung wird das Wägeglas verschlossen und so schnell wie möglich in den Exsikkator gestellt.
Wird ein anderes Gerät als der Filtertiegel verwendet, so trocknet man im Trockenofen, um das Trockengewicht der Fasern ohne Verlust zu bestimmen.
I.7.1.2. Kühlung
Alle Kühlvorgänge werden in dem neben der Waage aufgestellten Exsikkator ausreichend lange durchgeführt, um ein völliges Abkühlen der Wägegläser zu erreichen, wobei die Abkühldauer mindestens 2 Stunden beträgt.
I.7.1.3. Wägung
Nach dem Kühlen wird das Wägeglas innerhalb von zwei Minuten nach Herausnahme aus dem Exsikkator gewogen. Wägegenauigkeit 0,0002 g.
I.7.2. Verfahren
Man entnimmt aus der vorbehandelten Vorprobe eine Analysenprobe von mindestens 1 g Gewicht. Das Garn und die Gewebe werden in Längen von etwa 10 mm ausgeschnitten und soweit wie möglich zerlegt (zerschnitten). Die Analysenprobe wird in einem Wägeglas getrocknet, im Exsikkator gekühlt und gewogen. Die Probe wird in ein Glasgefäß gegeben, das im entsprechenden Teil der Gemeinschaftsmethode beschrieben ist, anschließend wird das Wägeglas sofort wieder gewogen und das Trockengewicht der Probe durch Differenzbildung ermittelt. Die Analyse wird gemäß den Angaben in dem entsprechenden Teil der Methode zu Ende geführt. Der Rückstand wird mikroskopisch geprüft, um festzustellen, ob durch die Behandlung die lösliche Faser völlig ausgesondert worden ist.
I.8. Berechnung und Ergebnisdarstellung
Das Gewicht des unlöslichen Bestandteils wird als Prozentsatz des Gesamtgewichts der im Gemisch enthaltenen Fasern ausgedrückt.Der Prozentsatz des löslichen Faseranteils ergibt sich aus der Differenz. Die Ergebnisberechnung erfolgt auf der Basis der Trockengewichte der reinen Fasern, wobei einmal vereinbarte Zuschläge, zum anderen Berichtigungsfaktoren zur Berücksichtigung der Verluste bei der Vorbehandlung und der Analyse angewendet werden. Diese Berechnungen erfolgen nach der unter Punkt 1.8.2 angegebenen Formel.
I.8.1. Berechnung des prozentualen Gewichtsanteils der trockenen und reinen unlöslichen Bestandteile ohne Berücksichtigung des Gewichtsverlusts der Fasern bei der Vorbehandlung:
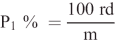
P1 ist der Prozentsatz des trockenen, reinen unlöslichen Faseranteils, m ist die Trockenmasse der Probe nach der Vorbehandlung,
r ist die Masse des trockenen Rückstands,
d ist der Berichtigungsfaktor zur Berücksichtigung des Gewichtsverlusts der unlöslichen Bestandteile im Reagenz bei der Analyse.
Geeignete Werte für "d" sind im entsprechenden Textteil der einzelnen Methoden anzugeben.
Selbstverständlich gelten die im Normalfall anwendbaren "d"-Werte nicht für chemisch angegriffene Fasern.
I.8.2. Berechnung des Prozentsatzes der unlöslichen Komponente nach Anwendung der vereinbarten Zuschläge und etwaiger Berichtigungsfaktoren zur Berücksichtigung des Gewichtsverlusts durch die Vorbehandlung

P1A % ist der Prozentsatz des unlöslichen Faseranteils unter Berücksichtigung des vereinbarten Zuschlags und des Gewichtsverlusts durch die Vorbehandlung.
P1 ist der Prozentsatz des unlöslichen, trockenen und reinen Faseranteils, errechnet aus der Formel nach 1.8.1,
a1 ist der vereinbarte Zuschlag für den unlöslichen Anteil (vgl. Anhang II der Richtlinie "Bezeichnung von Textilerzeugnisssen"),
a2 ist der vereinbarte Zuschlag des löslichen Faseranteils (vgl. Anhang II der Richtlinie "Bezeichnung von Textilerzeugnissen"),
b1 ist der prozentuale Gewichtsverlust des unlöslichen Faseranteils durch die Vorbehandlung,
b2 ist der prozentuale Gewichtsverlust des löslichen Faseranteils durch die Vorbehandlung.
Der Prozentsatz der zweiten Komponente (P2A%) beträgt 100 - P1A%.
Bei Anwendung einer besonderen Vorbehandlung müssen die Werte b1 und b2 nach Möglichkeit dadurch bestimmt werden, daß alle reinen Faserbestandteile der bei der Analyse angewandten Vorbehandlung unterworfen werden. Als reine Fasern gelten die Fasern, die frei von jeglichen nichtfaserhaltigen Stoffen sind, mit Ausnahme derjenigen Stoffe, die sie normalerweise (auf Grund ihrer Beschaffenheit und des Herstellungsprozesses) in dem Zustand (roh, gebleicht) enthalten, in dem sich die zu analysierende Ware befindet.
Verfügt man nicht getrennt über reine Faserbestandteile, die zur Herstellung der zu analysierenden Ware gedient haben, so sind für b1 und b2 Durchschnittswerte zugrunde zu legen, die aus der Prüfung ähnlicher Fasern ermittelt wurden, wie sie die untersuchte Mischung enthält.
Wird die normale Vorbehandlung durch Extraktion mit Petroläther und mit Wasser durchgeführt, so kann man im allgemeinen auf die Berichtigungsfaktoren b1 und b2 verzichten, außer im Fall von Rohbaumwolle, Rohflachs und Rohhanf, für die ein durch die Vorbehandlung bedingter Gewichtsverlust, von 4%, im Fall von Polypropylen ein solcher von 1% konventionell festgelegt ist.
Im Fall anderer Fasern wird konventionell festgelegt, daß der durch die Vorbehandlung bedingte Gewichtsverlust für die Berechnung unberücksichtigt bleibt.
II. Quantitative Analyse von Fasergemischen durch manuelle Trennung
II.1. Anwendungsbereich
Die Methode läßt sich auf Fasermischungen beliebiger Beschaffenheit anwenden, vorausgesetzt, daß sie keine untrennbare Mischung darstellen, und daß sie sich manuell trennen lassen.
II.2. Prinzip
Nach Identifizierung der einzelnen Bestandteile der Fasergemische werden zunächst die nicht faserhaltigen Bestandteile durch eine geeignete Vorbehandlung ausgesondert, anschließend die Fasern von Hand getrennt, getrocknet und zwecks Berechnung des Anteils der einzelnen Faserarten am Gemisch gewogen.
II.3. Erforderliche Geräte
II.3.1. Wägeglas bzw. andere Geräte, die gleichartige Ergebnisse liefern
II.3.2. Exsikkator mit gefärbtem Kieselgel als Feuchtigkeitsindikator
II.3.3. Trockenofen mit Ventilator zur Trocknung der Analysenproben bei (105 ± 3) °C
II.3.4. Analysenwaage, Empfindlichkeit 0,0002 g
II.3.5. Extraktionsapparat Soxhlet oder gleichwertige Apparatur
II.3.6. Nadel
II.3.7. Garndrehungszähler oder gleichwertige Apparatur
II.4. Reagenzien
II.4.1. Petroläther, nachdestilliert, Siedebereich 40-60 °C
II.4.2. Destilliertes oder entionisiertes Wasser
II.5. Konditionierungs- und Analysenatmosphäre
Vgl. Punkt 1.4
II.6. Vorprobe
Vgl. Punkt 1.5
II.7. Vorbehandlung der Probe
Vgl. Punkt 1.6
II.8. Analysengang
II.8.1. Analyse von Garnen
Eine Analysenprobe von mindestens 1 g wird aus einer vorbehandelten Probe entnommen. Bei sehr feinen Garnen kann die Analyse ungeachtet des Gewichts auf einer Mindestlänge von 30 m durchgeführt werden.
Die Garne sind in Stücke von geeigneter Länge zu schneiden; aus diesen sind die einzelnen Elemente mit Hilfe einer Präpariernadel und, falls erforderlich, mit Hilfe des Garndrehungszählers herauszutrennen. Die auf diese Weise herausgetrennten Elemente werden dann in ein tariertes Wägeglas gegeben und bei (105 ± 3) °C getrocknet, bis einn konstantes Gewicht gemäß 1.7.1 und 1.7.2 erreicht ist.
II.8.2. Analyseeines Gewebes
Eine Analysenprobe von mindestens 1 g wird aus einer vorbehandelten Probe entnommen, die Analysenprobe wird so ausgeschnitten, daß sie außerhalb der Webkante liegt, exakt geschnittene Ränder ohne Kräuselung aufweist und parallel zu Schuß und Kette bzw. bei Gewirken gleichlaufend längs und quer zu den Maschenreihen geschnitten ist. Die einzelnen Garne werden getrennt und in tarierten Wägegläsern gesammelt; dann wird wie unter Punkt II.8.1 vorgegangen.
II.9. Berechnung und Ergebnisdarstellung
Das Gewicht jedes Bestandteils wird als Prozentsatz des Gesamtgewichts der im Gemisch enthaltenen Fasern ausgedrückt. Die Berechnung erfolgt auf der Basis des Trockengewichts der reinen Fasern unter Anwendung von vereinbarten Zuschlägen sowie von Berichtigungsfaktoren zur Berücksichtigung der während der Vorbehandlung aufgetretenen Gewichtsverluste.
II.9.1. Berechnung des Prozentsatzes der reinen Trockengewichte ohne Berücksichtigung des Gewichtsverlusts der Fasern durch die Vorbehandlung.

P1 ist der Prozentsatz des trockenen und reinen ersten Faseranteils,
m1 ist die reine Trockenmasse des ersten Faseranteils,
m2 ist die reine Trockenmasse des zweiten Faseranteils.
II.9.2. Berechnung des Prozentsatzes jeder einzelnen Komponente nach Anwendung der vereinbarten Zuschläge und etwaiger Berichtigungsfaktoren zur Berücksichtigung des Gewichtsverlustes durch die Vorbehandlung: siehe Punkt I.8.2.
III. 1. Genauigkeit des Verfahrens
Die für jedes Verfahren angegebene Genauigkeit bezieht sich auf die Reproduzierbarkeit (Wiederholstreubereich).
Die Reproduzierbarkeit ist der Zuverlässigkeitsgrad, d. h. die Einengung der Übereinstimmung zwischen den Versuchsergebnissen, die in verschiedenen Laboratorien oder zu verschiedenen Zeiträumen erzielt werden, wenn dabei jeweils nach demselben Verfahren und an demselben homogenen Prüfgut Einzelergebnisse ermittelt werden.
Die Reproduzierbarkeit wird durch die Zuverlässigkeitsgrenzen der Versuchsergebnisse bei einer statistischen Sicherheit von 95 % ausgedrückt.
Dies besagt, daß die Abweichung zwischen den Ergebnissen einer in verschiedenen Laboratorien durchgeführten Analysenreihe bei richtiger und normaler Anwendung der Methode an einer gleichartigen homogenen Mischung nur in fünf von hundert Fällen überschritten werden darf.
III.2. Analysenbericht
III.2.1. Angabe, ob die Analyse nach dem hier beschriebenen Verfahren durchgeführt worden ist.
III.2.2. Detaillierte Angaben über etwaige Spezial-Vorbehandlungen (siehe Punkt I.6).
III.2.3. Angabe der Einzelergebnisse sowie des arithmetischen Mittels auf eine Dezimalstelle genau.
| Methode | Anwendungsbereich1 | Reagenz | |
| Löslicher Bestandteil | Unlöslicher Bestandteil | ||
| 1. | Acetat | Bestimmte andere Fasern | Aceton |
| 2. | Bestimmte Eiweißfasern | Bestimmte andere Fasern | Hypochlorit |
| 3. | Viskose, Cupro und bestimmte typen von Modal | Bestimmte andere Fasern | Ameisensäure und Zinkchlorid |
| 4. | Polyamid oder Nylon | Bestimmte andere Fasern | 80 %ige Ameisensäure |
| 5. | Acetat | Bestimmte andere Fasern | Benzylalkohol |
| 6. | Triacetat oder Polylactid | Bestimmte andere Fasern | Dichlormethan |
| 7. | Bestimmte Zellulosefasern | Bestimmte andere Fasern | 75 %ige Schwefelsäure |
| 8. | Polyacrylfasern, bestimmte Modacrylfasern oder bestimmte Polychloridfasern | Bestimmte andere Fasern | Dimethylformamid |
| 9. | Bestimmte Polychloridfasern | Bestimmte andere Fasern | Schwefelkohlenstoff/Aceton (55,5/44,5) |
| 10. | Acetat | Bestimmte andere Fasern | Essigsäure |
| 11. | Seide, Polyamid oder Nylon | Bestimmte andere Fasern | 75 %ige Schwefelsäure |
| 12. | Jute | Bestimmte Fasern tierischen Ursprungs | Stickstoffbestimmungsverfahren |
| 13. | Polypropylen | Bestimmte andere Fasern | Xylol |
| 14. | Bestimmte Fasern | Bestimmte andere Fasern | Konzentrierte Schwefelsäure |
| 15. | Polychloridfasern, bestimmte Modacryle, bestimmte Elastane, Acetate, Triacetate | Bestimmte andere Fasern | Cyclohexanon |
| 16. | Melamin | Bestimmte andere Fasern | 90 %ige heiße Ameisensäure |
| 1) Die Fasern sind unter der jeweiligen Methode genau aufgelistet. | |||
Verfahren Nr. 1 11
ACETAT- UND BESTIMMTE ANDERE FASERN
(Azeton-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
Selbstverständlich ist dieses Verfahren nicht auf Oberflächenentacetylierte Acetatfasern anwendbar.
2. GRUNDLAGE DES VERFAHRENS
Die Acetatfasern werden mittels Aceton aus einer bekannten Trockenmasse herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen, seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse einer Mischung ausgedrückt. Der Anteil an trockenen Acetatfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Gerät
200-ml-Erlenmeyerkolben mit Glasschliffstopfen
3.2. Reagenz
Aceton
4. DURCHFÜHRUNG
Es ist der im Allgemeinen Teil beschriebene Analysengang zu befolgen und dabei folgendermaßen vorzugehen:
Der Probe, die sich in einem mit einem Glasschliffstopfen versehenen Erlenmeyerkolben von mindestens 200 ml befindet, werden 100 ml Aceton je Gramm Probe zugegeben; der Kolben wird geschüttelt und 30 Minuten lang bei Raumtemperatur unter zeitweiligem Schütteln stehengelassen, anschließend wird die Flüssigkeit über einen gewogenen Glasfiltertiegel dekantiert.
Diese Behandlung wird noch zweimal wiederholt (insgesamt drei Extraktionen), doch jeweils nur 15 Minuten lang, so daß die Gesamtdauer der Acetonbehandlung eine Stunde beträgt. Der Rückstand wird in einen Glasfiltertiegel überführt und danach mit Aceton unter Absaugen ausgewaschen. Der Tiegel wird erneut mit Aceton gefüllt, das man ohne Absaugen von selbst ablaufen läßt.
Zum Schluß wird der Tiegel unter Absaugen entleert; Tiegel und Rückstand werden getrocknet, gekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im Allgemeinen Teil angegebenen Berechnungsverfahren berechnet. Der Wert "d" beträgt 1,00, ausgenommen bei Melamin: 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einer statistischen Sicherheit von 95 %.
Verfahren Nr. 2 11
BESTIMMTE EIWEISSFASERN UND BESTIMMTE ANDERE FASERN
(Hypochlorit-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
Sind unterschiedliche Eiweißfasern vorhanden, so liefert das Verfahren deren Gesamtmenge, jedoch nicht die prozentualen Anteile.
2. GRUNDLAGE DES VERFAHRENS
Die Eiweißfasern werden mit einer Hypochloritlösung aus einer bekannten Trockenmasse herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen, seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenen Eiweißfasern wird durch Differenzbildung ermittelt.
Für die Herstellung der Hydrochloritlösung kann Lithiumhypochlorit oder Natriumhypochlorit verwendet werden.
Lithiumhypochlorit empfiehlt sich dann, wenn die Zahl der Analysen gering ist oder Analysen in größeren zeitlichen Abständen durchgeführt werden. Der Grund liegt darin, daß festes Lithiumhypochlorit gegenüber Natriumhypochlorit einen nahezu konstanten Hypochloritanteil enthält. Ist dieser Hypochloritanteil bekannt, muß nicht bei jeder Analyse der Hypochloritgehalt jodometrisch überprüft werden, es kann vielmehr mit konstanter Einwaage an Lithiumhypochlorit gearbeitet werden.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Geräte
3.2. Reagenzien
Diese besteht aus einer frisch zubereiteten Lösung mit 35 (± 2) g/l aktivem Chlor (etwa 1 M), der 5 (± 0,5) g/l vorher gelöstes Natriumhydroxid zugegeben wurde. Man löst hierzu 100 g Lithiumhypochlorit mit 35 % aktivem Chlor (bzw. 115 g mit 30 % aktivem Chlor) in etwa 700 ml destilliertem Wasser, fügt 5 g in etwa 200 ml destilliertem Wasser gelöstes Natriumhydroxid hinzu und füllt auf 1 Liter auf. Die frisch hergestellte Lösung braucht nicht jodometrisch überprüft zu werden.
Diese besteht aus einer frisch zubereiteten Lösung, mit 35 (± 2) g/l aktivem Chlor (etwa 1 M), der 5 (± 0,5) g/l vorher gelöstes Natriumhydroxid zugegeben wurden.
Vor jeder Analyse ist der Gehalt der Lösung an aktivem Chlor jodometrisch zu überprüfen.
5 ml Eisessig werden mit Wasser auf 1 Liter aufgefüllt.
4. DURCHFÜHRUNG
Es ist der im Allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen: Etwa 1 g der Probe wird in den 250 ml-Kolben mit etwa 100 ml der Hypochloritlösung (Lithium- oder Natriumhypochlorit) versetzt und gut geschüttelt, um die Probe zu benetzen.
Anschließend wird der Kolben 40 Minuten in einem Thermostat bei 20 °C gestellt und dabei kontinuierlich oder zumindest häufig geschüttelt. Da die Lösung der Wolle exotherm verläuft, ist die Reaktionswärme durch diese Arbeitsweise zu verteilen und abzuführen. Andernfalls können größere Fehler durch das Anlösen der unlöslichen Fasern entstehen.
Nach 40 Minuten wird der Inhalt des Kolbens durch einen gewogenen Glasfiltertiegel filtriert; etwa zurückgebliebene Fasern werden durch Auswaschen des Kolbens mit etwas Hypochloritreagens in den Filtertiegel gespült. Der Filtertiegel wird mittels Unterdruck entleert und der Rückstand nacheinander mit Wasser, verdünnter Essigsäure und wieder mit Wasser gewaschen, wobei der Tiegel nach jeder Flüssigkeitszugabe unter Absaugen entleert wird, jedoch erst, nachdem die Flüssigkeit ohne Absaugen durchgelaufen ist.
Zum Schluß wird der Tiegel durch Absaugen geleert, zusammen mit dem Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im Allgemeinen Teil angegebenen Berechnungsverfahren berechnet. Der Wert "d" beträgt 1,00, für Baumwolle, Viskose, Modal und Melamin 1,01 und für ungebleichte Baumwolle 1,03.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einer statistischen Sicherheit von 95 %.
Verfahren Nr. 3 11
VISKOSE, CUPRO ODER GEWISSE typeN VON MODAL BESTIMMTE ANDERE FASERN
(Verfahren mit Ameisensäuren und Zinkchlorid)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
Wird die Anwesenheit von Modalfasern festgestellt, so ist ein Vorversuch auszuführen, um zu untersuchen, ob diese im Reagenz löslich sind.
Das Verfahren gilt nicht für Gemische, bei denen die Baumwolle durch übermäßigen chemischen Angriff verändert worden ist oder die Viskose- oder Cuprofasern durch das Vorhandensein bestimmter Farbstoffe, Reagenzien oder Appreturmittel, die nicht vollständig entfernt werden können, nicht mehr vollständig löslich sind.
2. GRUNDLAGE DES VERFAHRENS
Die Viskose, Cupro- oder Modalfasern werden aus einer bekannten Trockenmasse mit einem Reagenz aus Ameisensäure und Zinkchlorid herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen, seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenen Viskose-, Cupro- oder Modalfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Geräte
3.2. Reagenzien
Anmerkung
In diesem Zusammenhang wird auf Punkt 1.3.2.2 hingewiesen, der vorschreibt, daß alle verwendeten Reagenzien chemisch rein sein müssen; außerdem darf ausschließlich geschmolzenes wasserfreies Zinkchlorid verwendet werden.
4. DURCHFÜHRUNG
Es ist der im Allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen: Man gibt die Probe sofort in einen auf 40 °C vorgewärmten Erlenmeyer, versetzt sie mit 100 ml der Ameisensäure-Zinkchloridlösung je Gramm Probe, die auf 40° vorgewärmt ist. Der Kolben wird verschlossen und geschüttelt. Der Kolben und sein Inhalt werden 21/2 Stunden lang bei 40 °C stehengelassen und während dieser Zeit zweimal in Intervallen von je 1 Stunde geschüttelt. Der Inhalt des Kolbens wird über einen gewogenen Glasfiltertiegel filtriert; dabei werden etwa am Kolben haftende Fasern mit Reagenzlösung in den Filtertiegel gespült. Mit 20 ml Reagenz nachspülen.
Man wäscht Filtertiegel und Rückstand mit Wasser von 40 °C. Danach spült man den Faserrückstand mit ca. 100 ml kalter Ammoniaklösung (3.2 (ii) ), wobei sichergestellt werden muß, daß dieser Rückstand 10 Minuten lang vollständig in der Lösung eingetaucht bleibt 1; danach spült man gründlich mit kaltem Wasser.
Keinen Unterdruck anwenden, solange die Spülflüssigkeit nicht von selbst vollständig durchgelaufen ist. Zum Schluß wird der noch verbleibende Flüssigkeitsüberschuß durch Absaugen entfernt und Tiegel und Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Wert 'd' beträgt 1,00; nur für Baumwolle beträgt er 1,02 und für Melamin 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 2 mit einer statistischen Sicherheit von 95 %.
__________
1) Um die Einwirkungsdauer der Ammoniaklösung auf den Faserrückstand während 10 Minuten sicherzustellen, kann man z.B. den Filtertiegel mit einem Vorstoß mit Hahn versehen, der den Abfluß der Ammoniaklösung zu regeln gestattet.
Verfahren Nr. 4
POLYAMID ODER NYLON UND BESTIMMTE ANDERE FASERN
(Verfahren mit 80 %iger Ameisensäure)
1. Anwendungsbereich
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
Das Verfahren gilt wie vorstehend angegeben für wollhaltige Mischungen, doch ist bei einem Wollgehalt von über 25 % das Verfahren Nr. 2 anzuwenden, d. h. Auflösung der Wolle in einer Natriumhypochlorit-Lösung.
2. Grundlage des Verfahrens
Das Polyamid wird aus einer bekannten Trockenmasse mittels Ameisensäure herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen, seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenem Polyamid oder Nylon wird durch Differenzbildung ermittelt.
3. Geräte und Reagenzien (neben den im Allgemeinen Teil genannten)
3.1. Gerät
200-ml-Erlenmeyerkolben mit Glasschliffstopfen
3.2. Reagenzien
Zwischen 77 und 83 Gewichtsprozent Ameisensäure ist die Konzentration nicht kritisch.
4. DURCHFÜHRUNG
Es ist der im Allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Die Probe wird mit einem 200-ml-Erlenmeyerkolben mit 100 ml Ameisensäure je Gramm Probe versetzt, der Kolben wird verschlossen und geschüttelt, um die Probe vollständig zu benetzen; 15 Minuten bei Raumtemperatur unter zeitweiligem Schütteln stehenlassen. Der Inhalt des Erlenmeyerkolbens wird durch einen gewogenen Glasfiltertiegel filtriert; etwa zurückbleibende Fasern werden durch Auswaschen des Kolbens mit etwas Ameisensäurelösung in den Filtertiegel überführt. Der Filtertiegel wird unter Absaugen entleert und der Rückstand nacheinander mit Ameisensäure, warmem Wasser, verdünntem Ammoniak und schließlich mit kaltem Wasser gewaschen, wobei der Tiegel nach jeder Flüssigkeitszugabe unter Absaugen entleert wird, jedoch erst, nachdem die Flüssigkeit ohne Absaugen durchgelaufen ist. Zum Schluß wird der Tiegel durch Absaugen geleert, zusammen mit dem Rückstand getrocknet, gekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im Allgemeinen Teil angegebenen Berechnungsverfahren berechnet. Der Wert "d" beträgt 1,00, ausgenommen bei Melamin: 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einer statistischen Sicherheit von 95 %.
Verfahren Nr. 5 11
ACETATFASERN UND BESTIMMTE ANDERE FASERN
(Benzylalkohol-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
2. GRUNDLAGE DES VERFAHRENS
Die Acetatfasern werden aus einer bekannten Trockenmasse des Gemischs mit Hilfe von Benzylalkohol von 52 ± 2 °C herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenem Acetat wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Geräte
3.2. Reagenzien
4. DURCHFÜHRUNG
Es ist den im Allgemeinen Teil gegebenen Anweisungen zu folgen und folgendermaßen vorzugehen:
Der im Kolben befindlichen Probe werden 100 ml Benzylalkohol je Gramm Probe zugegeben. Der Kolben wird mit einem Stopfen verschlossen und so auf einem Schüttler befestigt, daß er in ein Wasserbad von 52 ± 2 °C taucht, wo er mindestens 20 Minuten geschüttelt wird. (Unter Umständen kann der mechanische Schüttler durch kräftiges Schütteln mit der Hand ersetzt werden.)
Die Flüssigkeit wird über einen gewogenen Glasfiltertiegel dekantiert. Eine neue Portion Benzylalkohol wird hinzugefügt und der Kolben nochmals 20 Minuten lang bei 52 ± 2 °C geschüttelt. Dann wird über den Filtertiegel dekantiert. Diese Behandlung wird ein drittes Mal wiederholt.
Danach wird die Flüssigkeit und der Rückstand in den Filtertiegel gegossen. Etwa im Kolben zurückbleibende Fasern werden durch Hinzufügung einer zusätzlichen Menge Benzylalkohol von 52 ± 2 °C übergespült.
Der Tiegel wird nun vollständig trockengeschleudert.
Die Fasern werden in einen Kolben überführt; Äthylalkohol wird zum Spülen beigefügt. Nach kräftigem Schütteln mit der Hand wird über den Filtertiegel dekantiert.
Dieser Spülvorgang ist zwei- oder dreimal zu wiederholen. Der Rückstand wird in den Tiegel überführt und vollständig trockengeschleudert. Filtertiegel und Rückstand werden getrocknet und nach Abkühlung gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im Allgemeinen Teil angegebenen Berechnungsverfahren berechnet. Der Wert "d" beträgt 1,00, ausgenommen bei Melamin: 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenem Textilfasergemisch liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einer statistischen Sicherheit von 95 %.
Verfahren Nr. 6 11
TRIACETAT- POLYLACTIDFASERN UND BESTIMMTE ANDERE FASERN
(Dichlormethan-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
Anmerkung:
Triacetatfasern, die durch besondere Behandlung partiell verseift sind, sind im Reagenz nicht mehr voll löslich. In diesem Fall ist das Verfahren nicht anwendbar.
2. GRUNDLAGE DES VERFAHRENS
Die Triacetat- oder Polylactidfasern werden aus einer bekannten Trockenmasse mit Hilfe von Dichlormethan herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenem Triacetat oder Polylactid wird durch Differenzbildung erreicht.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Gerät
200-ml-Erlenmeyerkolben mit Glasschliffstopfen
3.2. Reagenz
4. DURCHFÜHRUNG
Es ist der im Allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Die in einem 200-ml-Erlenmeyerkolben mit Schliffstopfen befindliche Probe wird mit 100 ml Dichlormethan je Gramm Probe versetzt, der Kolben wird mit dem Stopfen verschlossen, in Abständen von zehn Minuten kräftig geschüttelt zwecks vollständiger Benetzung der Probe und 30 Min. bei Raumtemperatur unter zeitweiligem, regelmäßigem Schütteln stehengelassen. Die Flüssigkeit wird durch einen gewogenen Glasfiltertiegel dekantiert. Man gibt 60 ml Dichlormethan in den Kolben mit dem Rückstand, schüttelt von Hand und filtriert den Inhalt des Kolbens über den Glasfiltertiegel. Etwa zurückbleibende Fasern werden durch Spülen mit einer kleinen zusätzlichen Menge von Dichlormethan in den Tiegel überführt. Der Tiegel wird unter Absaugen entleert, dann erneut mit Dichlormethan gefüllt, das man vollständig ablaufen läßt.
Schließlich wird der Flüssigkeitsüberschuß unter Absaugen entfernt, der Rückstand zur gänzlichen Entfernung des Lösungsmittels mit kochendem Wasser behandelt, abgesaugt und der Tiegel mit Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im Allgemeinen Teil angegebenen Berechnungsverfahren berechnet. Der Wert "d" beträgt 1,00, ausgenommen bei Polyester, Elastomultiester, Elastolefin und Melamin: 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einer statistischen Sicherheit von 95 %.
Verfahren Nr. 7 11
BESTIMMTE ZELLULOSEFASERN UND BESTIMMTE ANDERE FASERN
(Verfahren mit 75 %iger Schwefelsäure)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
2. GRUNDLAGE DES VERFAHRENS
Die Zellulosefasern werden aus einer bekannten Trockenmasse mit Hilfe 75 %iger Schwefelsäure herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenen Zellulosefasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Geräte
3.2. Reagenzien
Herstellung in der Weise, daß 700 ml Schwefelsäure, Dichte bei 20 °C: 1,84 vorsichtig zu 350 ml destilliertem Wasser zugesetzt werden. Nach Abkühlung auf Raumtemperatur mit Wasser auf 1 l auffüllen.
80 ml Ammoniaklösung, Dichte bei 20°C: 0,88, werden mit Wasser auf 1 l aufgefüllt.
4. DURCHFÜHRUNG
Es ist nach den im Allgemeinen Teil gegebenen Anweisungen zu arbeiten und folgendermaßen vorzugehen:
Die in einem 500-ml-Erlenmeyerkolben mit Schliffstopfen befindliche Probe wird mit 200 ml 75 %iger Schwefelsäure je Gramm Probe versetzt, der Kolben wird mit dem Stopfen verschlossen und vorsichtig geschüttelt, um die Probe vollständig zu benetzen. Der Kolben wird eine Stunde lang auf einer Temperatur von 50 ± 5 °C gehalten und in Abständen von etwa 10 Minuten geschüttelt. Danach wird der Inhalt des Kolbens unter Absaugen durch einen Filtertiegel filtriert. Etwa zurückgebliebene Fasern werden durch Spülen des Kolbens mit etwas 75 %iger Schwefelsäure in den Glasfiltertiegel überführt. Der Glasfiltertiegel wird durch Absaugen geleert und der auf dem Filter verbliebene Rückstand durch erneute Zugabe von 75 %iger Schwefelsäure ein erstes Mal ausgewaschen. Es ist erst abzusaugen, nachdem die Flüssigkeit ohne Absaugen hindurchgelaufen ist. Der Rückstand wird nacheinander mehrmals mit kaltem Wasser, zweimal mit verdünnter Ammoniaklösung und dann gründlich mit kaltem Wasser gewaschen, wobei nach jeder Flüssigkeitszugabe abzusaugen ist, jedoch jedesmal erst, nachdem die Flüssigkeit ohne Absaugen hindurchgelaufen ist. Schließlich wird der Tiegel durch Absaugen entleert, zusammen mit dem Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Wert 'd' beträgt 1,00; nur für Polypropylen/Polyamid-Bikomponentenfasern beträgt er 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einer statistischen Sicherheit von 95 %.
Verfahren Nr. 8 11
POLYACRYLFASERN, MODACRYLFASERN,BESTIMMTE POLYCHLORVINYLFASERN UND BESTIMMTE ANDERE FASERN
(Dimethylformamid-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
Es gilt ferner für Polyacryl- und bestimmte Modacrylfasern, die mit vormetallisierten Farbstoffen, jedoch nicht mit Nachchromierfarbstoffen behandelt sind.
2. GRUNDLAGE DES VERFAHRENS
Die Polyacrylfasern, bestimmte Modacrylfasern oder bestimmte Polychlorvinylfasern werden aus einer bekannten Trockenmasse mittels Dimethylformamid im kochenden Wasserbad herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen, seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenen Polyacrylfasern, Modacrylfasern oder Polychlorvinylfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Geräte
3.2. Reagenz
Dimethylformamid (Siedepunkt 153 ± 1) °C mit nicht mehr als 1 % Wasser. Da das Reagenz giftig ist, wird empfohlen, unter dem Abzug zu arbeiten.
4. DURCHFÜHRUNG
Es ist nach den im Allgemeinen Teil angegebenen Anweisungen zu arbeiten und folgendermaßen vorzugehen:
Die in einem 200-ml-Erlenmeyerkolben mit Schliffstopfen befindliche Probe wird mit 80 ml im kochenden Wasserbad vorgewärmtem Dimethylformamid je Gramm Probe versetzt, der Kolben mit dem Stopfen verschlossen, geschüttelt zwecks vollständiger Benetzung der Probe und eine Stunde lang im kochenden Wasserbad belassen. Während dieser Zeit wird der Kolben und sein Inhalt fünfmal vorsichtig geschüttelt.
Die Flüssigkeit wird durch einen gewogenen Glasfiltertiegel dekantiert, wobei die Fasern im Erlenmeyer zurückgehalten werden. Man gibt erneut 60 ml Dimethylformamid in den Kolben und erwärmt wiederum 30 Minuten im kochenden Wasserbad, wobei der Kolben und sein Inhalt zweimal vorsichtig von Hand geschüttelt werden.
Der Inhalt des Kolbens wird durch einen Glasfiltertiegel unter Absaugen filtriert.
Die zurückbleibenden Fasern werden durch Ausspülen des Kolbens mit Dimethylformamid in den Glasfiltertiegel überführt. Der Tiegel wird unter Absaugen entleert. Die zurückbleibenden Fasern werden mit etwa 1 Liter Wasser von 70-80 °C Temperatur gewaschen, wobei der Tiegel jedesmal mit Wasser gefüllt wird. Nach jeder Wasserzugabe wird kurz abgesaugt, allerdings erst dann, wenn das Wasser abgelaufen ist. Läuft das Waschwasser zu langsam durch den Tiegel, kann kurz abgesaugt werden.
Zum Schluß wird der Tiegel mit dem Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im Allgemeinen Teil angegebenen Berechnungsverfahren berechnet. Der Wert "d" beträgt 1,00, ausgenommen bei Wolle, Baumwolle, Cupro, Modal, Polyester, Elastomultiester und Melamin: 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei max. ± 1 mit einer statistischen Sicherheit von 95 %.
___________
1) Die Löslichkeit dieser Modacryl- oder Polychlorvinylfasern im Reagenz ist vor der Analyse zu prüfen.
Verfahren Nr. 9 11
BESTIMMTE POLYCHLORVINYLFASERN UND BESTIMMTE ANDERE FASERN
(Verfahren mit Schwefelkohlenstoff/Aceton 55,5/44,5)
1. ANWENDUNGSBEREICH
Das Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
Ist der Gehalt an Wolle oder Seide im Gemisch größer als 25 %, so ist Verfahren Nr. 2 anzuwenden.
Ist der Gehalt an Polyamid oder Nylon im Gemisch größer als 25 %, so ist Verfahren Nr. 4 anzuwenden.
2. GRUNDLAGE DES VERFAHRENS
Die Polychloridfasern werden aus einer bekannten Trockenmasse mit Hilfe einer aseotropischen Mischung von Schwefelkohlenstoff und Aceton herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen, seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenen Polychlorvinylfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Geräte
3.2. Reagenzien
Da das Reagenz giftig ist, wird empfohlen, unter dem Abzug zu arbeiten
4. DURCHFÜHRUNG
Es ist nach den im Allgemeinen Teil gegebenen Anweisungen zu arbeiten und folgendermaßen vorzugehen:
Die in einem 200-ml-Erlenmeyerkolben mit Schliffstopfen befindliche Probe wird mit 100 ml aseotropischer Mischung je Gramm Probe versetzt. Der Kolben wird sorgfältig verschlossen und 20 Minuten lang mit dem mechanischen Schüttler oder von Hand kräftig geschüttelt. Die Flüssigkeit wird durch einen gewogenen Glasfiltertiegel dekantiert.
Die Behandlung wird mit 100 ml frischem Lösungsmittel wiederholt. Diese Behandlung wird so oft fortgesetzt, bis ein Tropfen Lösungsmittel nach Verdunstung auf einem Uhrglas keinerlei Polymerniederschlag hinterläßt. Der Rückstand wird mit Hilfe einer zusätzlichen Menge Lösungsmittel in den Filtertiegel überführt, der Filtertiegel durch Sauganwendung entleert; Tiegel und Rückstand werden mit 20 ml Alkohol gespült und anschließend dreimal mit Wasser nachgespült. Vor dem Absaugen muß die Spülflüssigkeit vollständig durchgelaufen sein. Tiegel und Rückstand werden getrocknet, abgekühlt und gewogen.
Anmerkung:
Die Proben mancher Mischungen mit hohem Gehalt an Polyvinylchlorid schrumpfen beim Trocknen beträchtlich, was die Beseitigung des Polyvinylchlorids durch das Lösungsmittel erschwert. Jedoch verhindert diese Schrumpfung nicht die vollständige Auflösung des Polyvinylchlorids.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im Allgemeinen Teil angegebenen Berechnungsverfahren berechnet. Der Wert "d" beträgt 1,00, ausgenommen bei Melamin: 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einer statistischen Sicherheit von 95 %.
_______
1) Die Löslichkeit der Polychlorvinylfasern im Reagenz muß überprüft werden, bevor die Analyse durchgeführt wird.
Verfahren Nr. 10 11
ACETATFASERN UND BESTIMMTE ANDERE FASERN
(Essigsäure-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
2. GRUNDLAGE DES VERFAHRENS
Die Acetatfasern werden aus einer bekannten Trockenmasse mittels Essigsäure aufgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenen Acetatfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Geräte
3.2. Reagenz
Essigsäure (mehr als 99 %). Dieses Reagenz ist sehr ätzend, und bei seiner Verwendung ist daher Vorsicht geboten.
4. DURCHFÜHRUNG
Es ist nach den im Allgemeinen Teil gegebenen Anweisungen zu arbeiten und folgendermaßen vorzugehen:
Die in einem 200-ml-Erlenmeyerkolben mit Schliffstopfen befindliche Probe wird mit 100 ml Essigsäure je Gramm Probe versetzt. Der Kolben wird sorgfältig verschlossen und bei Raumtemperatur 20 Minuten mit einem mechanischen Schüttler oder von Hand kräftig geschüttelt. Die Flüssigkeit wird durch den gewogenen Glasfiltertiegel dekantiert. Diese Behandlung wird zweimal unter Verwendung von jeweils 100 ml frischem Lösungsmittel wiederholt, so daß insgesamt drei Extraktionen ausgeführt werden. Der Rückstand wird in den Filtertiegel überführt, der Flüssigkeitsrückstand unter Sauganwendung entleert; Tiegel und Rückstand werden mit 50 ml Essigsäure sowie anschließend dreimal mit Wasser nachgespült. Nach jedem Auswaschen muß die Flüssigkeit von selbst durchlaufen, bevor abgesaugt wird. Tiegel und Rückstand trocknen, abkühlen und wägen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden in der im Allgemeinen Teil angegebenen Weise berechnet. Der Berichtigungsfaktor "d" beträgt 1,00.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einer statistischen Sicherheit von 95 %.
Verfahren Nr. 11 11
SEIDE ODER POLYAMID UND BESTIMMTE ANDERE FASERN
(Verfahren mit 75 %iger Schwefelsäure)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
2. GRUNDLAGE DES VERFAHRENS
Die Seidenfasern bzw. Polyamid- oder Nylonfasern werden aus einer bekannten Trockenmasse mit 75-gewichtsprozentiger Schwefelsäure herausgelöst.
Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen. Seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse des Gemischs ausgedrückt. Der Anteil an trockener Seide bzw. trockenem Polyamid oder Nylon wird durch Differenzbildung ermittelt
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Gerät
200-ml-Erlenmeyerkolben mit Schliffstopfen
3.2. Reagenzien
Herstellungsweise: 700 ml Schwefelsäure, Dichte 1,84 bei 20 °C, werden vorsichtig unter Abkühlung zu 350 ml destilliertem Wasser zugesetzt.
Nach Abkühlung auf Raumtemperatur mit Wasser auf 1 l auffüllen.
4. DURCHFÜHRUNG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Die in einem 200-ml-Erlenmeyerkolben mit Schliffstopfen befindliche Probe wird mit 100 ml 75 %iger Schwefelsäure je Gramm Probe versetzt. Der Kolben wird verschlossen, kräftig geschüttelt und 30 Minuten bei Raumtemperatur stehen gelassen. Erneut schütteln, 30 Minuten stehen lassen. Ein letztes Mal schütteln und den Inhalt des Kolbens in den gewogenen Glasfiltertiegel überführen. Etwa im Kolben zurückbleibende Fasern werden mit 75 %iger Schwefelsäure nachgespült. Der Rückstand wird im Filtertiegel nacheinander mit 50 ml verdünnter Schwefelsäure, 50 ml Wasser und 50 ml verdünntem Ammoniak ausgewaschen. Die Fasern müssen etwa 10 Minuten lang mit der Flüssigkeit in Kontakt bleiben, bevor abgesaugt wird. Schließlich wird mit Wasser nachgewaschen, wobei die Fasern 30 Minuten im Wasser verbleiben. Die Restflüssigkeit wird unter Absaugen entfernt. Tiegel und Rückstand werden getrocknet, abgekühlt und gewogen.
Im Fall von binären Gemischen von Polyamid mit Polypropylen/Polyamid-Bikomponentenfasern ist nach dem Überführen der Fasern in den gewogenen Glasfiltertiegel und vor Anwendung des beschriebenen Waschverfahrens der Rückstand im Filtertiegel zweimal jeweils mit 50 ml 75%iger Schwefelsäure auszuwaschen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Wert 'd' beträgt 1,00; nur für Wolle beträgt er 0,985, für Polypropylen/Polyamid-Bikomponentenfasern 1,005 und für Melamin 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der Ergebnisse dieses Verfahrens bei maximal ± 1 mit einer statistischen Sicherheit von 95 %; nur bei binären Gemischen von Polyamid mit Polypropylen/Polyamid-Bikomponentenfasern liegen die Zuverlässigkeitsgrenzen bei maximal ± 2.
Verfahren Nr. 12
JUTE UND BESTIMMTE FASERN TIERISCHEN URSPRUNGS
(Verfahren mittels Stickstoffbestimmung)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
Letztere können aus Tierhaaren (2 und 3) oder aus Wolle (1) oder einer Mischung von Tierhaaren und Wolle bestehen. Selbstverständlich eignet sich das Verfahren nicht für Textilfasermischungen, die nichtfaserige Bestandteile (Farbstoffe, Appreturmittel usw.) auf Stickstoffbasis enthalten.
2. GRUNDLAGE DES VERFAHRENS
Man bestimmt den Stickstoffgehalt der Mischung und berechnet hieraus sowie aus dem bekannten Stickstoffgehalt der beiden Bestandteile der Mischung deren proportionalen Anteil.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Gerät
3.2. Reagenzien
4. VORBEHANDLUNG DER VORPROBE
An Stelle der im Allgemeinen Teil beschriebenen Vorbehandlung wird wie folgt vorgegangen:
Die lufttrockene Probe wird in einem Soxhlet-Apparat mit einer Mischung von einem Volumenteil Toluol und drei Volumenteilen Methanol 4 Stunden lang bei mindestens 5 Umläufen pro Stunde extrahiert. Man läßt die Lösungsmittel aus der Probe in Luft verdampfen und entfernt die letzten Spuren in einem Wärmeschrank bei 105 ± 3 °C. Sodann wird die Probe im Wasser (50 ml/g Probemenge) durch Sieden mit Rückkühlung 30 Minuten lang extrahiert. Nach dem Filtrieren wird die Probe in den Kolben zurückgegeben und die Extraktion mit einem gleichen Volumen frischen Wassers wiederholt. Nach erneutem Filtrieren wird das überschüssige Wasser durch Ausdrücken, Absaugen oder Zentrifugieren aus der Probe entfernt, die man dann an der Luft trocknen läßt.
Anmerkung:
Toluol und Methanol sind giftig; deshalb sind bei ihrer Verwendung alle erforderlichen Vorsichtsmaßnahmen zu treffen.
5. ANALYSIERUNG
5.1. Allgemeine Anweisung
Hinsichtlich Entnahme, Trocknung und Wägung der Probe sind die im Allgemeinen Teil gemachten Angaben zu befolgen.
5.2. Ausführliche Anleitung
Zu der Probe von mindestens 1 g im Kjeldahl-Kolben werden der Reihe nach 2,5 g Kaliumsulfat, 0,1 bis 0,2 g Selendioxyd und 10 ml Schwefelsäure (d = 1,84) gegeben. Der Kolben wird zunächst schwach erhitzt, bis das ganze Fasermaterial zersetzt ist, sodann wird kräftiger erhitzt, bis die Lösung klar und nahezu farblos geworden ist. Die Erhitzung wird 15 Minuten lang fortgesetzt, dann läßt man den Kolben abkühlen, verdünnt den Inhalt vorsichtig mit 10 bis 20 ml Wasser, kühlt ab, überführt den Inhalt quantitativ in einem 200-ml-Maßkolben und füllt das Volumen mit Wasser auf, um die Analysenlösung herzustellen.
In einen 100-ml-Erlenmeyerkolben werden etwa 20 ml Borsäurelösung gegeben; der Kolben wird unter dem Kondensator des Kjeldahl-Destilliergeräts so aufgestellt, daß das Ablaufrohr gerade unter den Spiegel der Borsäurelösung eintaucht. Es werden genau 10 ml Analysenlösung in den Destillierkolben überführt und mindestens 5 ml Natriumhydroxydlösung in den Trichter gegeben. Man hebt den Stopfen leicht an und läßt die Natriumhydroxydlösung langsam in den Kolben fließen. Wenn die Analysenlösung und die Natriumhydroxydlösung bestrebt sind, zwei getrennte Schichten zu bilden, so sind sie durch vorsichtiges Schütteln zu vermischen. Der Destillierkolben wird leicht erwärmt und gleichzeitig Dampf aus dem Dampferzeuger in ihn eingeführt. Es werden etwa 20 ml Destillat gesammelt und das Auffanggefäß so weit gesenkt, daß sich das Ende des Ablaufrohrs etwa 20 mm über dem Spiegel der Flüssigkeit befindet, danach wird eine Minute lang weiter destilliert. Das Ende des Ablaufrohrs wird mit Wasser ausgespült und das Reinigungswasser im Erlenmeyer aufgefangen. Der Erlenmeyer wird sodann entfernt und durch einen zweiten Erlenmeyer ersetzt, der etwa 10 ml Borsäurelösung enthält; darin werden etwa 10 ml Destillat aufgesammelt.
Die beiden Destillate werden getrennt mit 0,02-N-Schwefelsäure unter Verwendung der Indikatormischung titriert. Die Ergebnisse für die beiden Destillate werden notiert. Wenn das Bestimmungsergebnis des zweiten Destillats über 0,2 ml liegt, so ist die Bestimmung zu wiederholen; es muß also nochmals mit einem anderen aliquoten Teil der Analysenlösung destilliert werden.
Es wird ein Blindversuch durchgeführt, wobei die Zersetzung und Destillation lediglich unter Verwendung der Reagenzien erfolgt.
6. BERECHNUNG UND ERGEBNISDARSTELLUNG
6.1 Der prozentuale Anteil des Stickstoffs in der trockenen Probe wird folgendermaßen berechnet:
| 28 (V - b) N | |
| A% = | |
| W |
a % = Prozentsatz Stickstoff im reinen Trockengewicht der Probe
V = Gesamtvolumen in ml der Standardschwefelsäure für die Bestimmung
b = Gesamtvolumen in ml der Standardschwefelsäure beim Blindversuch
N = tatsächlicher Titel der Standardschwefelsäure
W = Trockenmasse (g) der Testprobe.
6.2. Bei Verwendung des Wertes 0,22 % für den Stickstoffgehalt der Jute bzw. 16,2 % für den Stickstoffgehalt der tierischen Fasern - beide
Werte auf die Trockenmasse bezogen - berechnet sich die Zusammensetzung der Mischung nach folgender Formel:
| a - 0,22 | ||
| PA% = | x 100 | |
| 16,2 - 0,22 |
PA % = Prozentsatz der tierischen Faser im reinen Trockengewicht der Probe.
7. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegen die Zuverlässigkeitsgrenzen der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einer statistischen Sicherheit von 95 %.
Verfahren Nr. 13
POLYPROPYLEN UND BESTIMMTE ANDERE FASERN
(Xylol-Verfahren)
1. ANWENDUNGSBEREICH
Diese Methode gilt nach Abtrennung der nichtfaserigen Bestandteile auf binäre Textilfasermischungen von:
mit
2. GRUNDLAGE DES VERFAHRENS
Die Polypropylenfaser wird aus einer bekannten Menge der Trockenmasse durch Lösung mit kochendem Xylol abgetrennt. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine Menge wird - gegebenenfalls nach der erforderlichen Korrektur - durch den Prozentanteil an der Trockenmasse ausgedrückt. Das Mengenverhältnis des Polypropylen wird aus der Gewichtsdifferenz berechnet.
3. GERÄTE UND REAGENZIEN (soweit nicht im Allgemeinen Teil erwähnt)
3.1. Geräte
3.2. Reagenz
Xylol, Siedebereich zwischen 137° und 14 °C.
Bemerkung:
Dieses Reagenz ist sehr leicht entflammbar und entwickelt giftige Dämpfe. Während der Verwendung sind deshalb entsprechende Vorsichtsmaßnahmen zu treffen.
4. DURCHFÜHRUNG
Es ist der im Allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Zu der in den Erlenmeyerkolben (3.1 i) ) eingegebenen Probe werden 100 ml Xylol (3.2) pro Gramm Probe hinzugegeben. Dann wird der Kühler (3.1 ii) ) aufgesetzt und der Kolbeninhalt zum Kochen gebracht; Kochdauer 3 Minuten. Die heiße Flüssigkeit wird sofort danach über den tarierten Glasfrittetiegel (siehe Bemerkung 1) dekantiert. Dieser Vorgang wird zweimal unter Verwendung von jeweils 50 ml frischem Lösungsmittel wiederholt.
Der im Kolben zurückgebliebene Rückstand wird nacheinander mit 30 ml kochendem Xylol (zweimal), dann zweimal mit je 75 ml Petroläther (siehe I 3.2.1 Allgemeiner Teil) ausgewaschen. Nach dem zweiten Waschen mit Petroläther wird der Inhalt des Kolbens durch den Tiegel filtriert; die zurückgebliebenen Fasern weden mittels einer kleinen zusätzlichen Menge Petroläther in den Tiegel überführt. Dann läßt man das Lösungsmittel vollständig verdampfen. Der Tiegel und der Rückstand werden getrocknet, abgekühlt und gewogen.
Bemerkungen:
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im Allgemeinen Teil angegebenen Berechnungsverfahren berechnet. Der Wert "d" beträgt 1,00, ausgenommen bei Melamin: 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei Anwendung dieses Verfahrens auf eine homogene Mischung von Textilstoffen liegt der Zuverlässigkeitsbereich der nach diesem Verfahren ermittelten Ergebnisse innerhalb von höchstens ± 1 bei einer Zuverlässigkeitsschwelle von 95 %.
________
1) Diesbezüglich sei auf die in "Melliand Textilberichte" 56 (1975), S. 643-645, beschriebene Apparatur hingewiesen.
Verfahren Nr. 14 11
BESTIMMTE FASERN UND BESTIMMTE ANDERE fASERN
(Verfahren mit konzentrierter Schwefelsäure)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
Die betreffenden Modacrylfasern sind diejenigen, die bei Behandlung in konzentrierter Schwefelsäure (d 20 = 1,84 g/ml) eine klare Lösung ergeben.
Dieses Verfahren kann insbesondere anstelle der Methoden Nr. 8 und 9 angewendet werden.
2. GRUNDLAGE DES VERFAHRENS
Alle Bestandteile außer Polychloridfasern, Polypropylen, Elastolefin, Melamin oder Polypropylen/Polyamid- Bikomponentenfasern (d. h. die unter 1.1 genannten Fasern) werden aus einer bekannten Trockenmasse durch Auflösen in konzentrierter Schwefelsäure (d20 = 1,84 g/ml) abgetrennt. Der aus Polychloridfasern, Polypropylen, Elastolefin, Melamin oder Polypropylen/Polyamid-Bikomponentenfasern bestehende Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil der anderen Bestandteile wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (soweit nicht im Allgemeinen Teil aufgeführt)
3.1. Geräte
3.2. Reagenzien
Um dieses Reagenz herzustellen, werden vorsichtig und unter Kühlung 400 ml Schwefelsäure (d20 = 1,84 g/ml) zu 500 ml Wasser hinzugegeben. Wenn die Lösung auf Zimmertemperatur abgekühlt ist, wird mit Wasser auf 1 Liter angefüllt;
60 ml einer konzentrierten Ammoniaklösung (d20 = 0,880 g/ml) werden mit destilliertem Wasser zu 1 Liter Lösung verdünnt.
4. DURCHFÜHRUNG
Es ist der im Allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Zu der in den Erlenmeyerkolben ( 3.1 i) ) eingegebenen Probe werden 100 ml Schwefelsäure (3.2 i) ) pro Gramm Probe hinzugegeben.
10 Minuten lang bei Zimmertemperatur unter öfterem Umrühren der Probenlösung mit dem Glasstab stehenlassen. Wenn es sich um ein Gewebe oder um gewirkte Ware handelt, so drückt man das Probestück leicht gegen die Kolbenwand, um auf diese Weise die durch die Schwefelsäure ausgelösten Bestandteile abzutrennen.
Die Flüssigkeit wird über den tarierten Glasfrittetiegel dekantiert. Dann werden erneut 100 ml Schwefelsäure ( 3.2. i) ) in den Kolben gegeben und der Vorgang wiederholt. Den Inhalt des Kolbens über dem Tiegel entleeren und hierbei den faserigen Rückstand mit dem Glasstab abtrennen. Erforderlichenfalls etwas konzentrierte Schwefelsäure (3.2. i) ) in den Kolben nachgeben, um die an den Wänden hängenbleibenden Faserreste zu erhalten. Den Tiegel durch Absaugen entleeren; das Filtrat des Kolbens völlig entfernen oder den Kolben auswechseln, dann den Rückstand im Tiegel nacheinander mit der 50 %igen Schwefelsäure (3.2. ii) ) destilliertem oder entionisiertem Wasser ( 1.3.2.3 Allgemeiner Teil) und der Ammoniaklösung ( 3.2. iii) ) und schließlich gründlich mit destilliertem oder entionisiertem Wasserauswaschen, wobei der Tiegel durch Absaugen nach jeder Zugabe vollständig entleert wird (während des Waschvorgangs ist nicht abzusaugen, sondern erst, nachdem die Flüssigkeit durch ihr Eigengewicht abgelaufen ist.)
Den Tiegel und den Rückstand trocknen, abkühlen und wiegen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Wert 'd' beträgt 1,00; nur für Melamin und Polypropylen/Polyamid-Bikomponentenfasern beträgt er 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegen die Zuverlässigkeitsgrenzen der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei die statistische Sicherheit 95 % beträgt.
Verfahren Nr. 15
POLYCHLORIDFASERN, BESTIMMTE MODACRYLE, BESTIMMTE ELASTHANE, ACETAT, TRIACETAT UND BESTIMMTE ANDERE FASERN
(Cyclohexanonverfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
Sind Modacryl- oder Elasthanfasern vorhanden, so ist ein Vorversuch notwendig, um festzustellen, ob die Fasern in dem Reagenz vollständig löslich sind.
Zur Bestimmung von Gemischen mit Polychloridfasern sind auch die Verfahren Nr. 9 oder 14 anwendbar.
2. GRUNDLAGE DES VERFAHRENS
Ausgehend von einer bekannten Trockenmasse des Gemischs werden Fasern von Acetat, Triacetat, Polychlorid, bestimmte Modacryle, bestimmte Elasthane, mit Cyclohexanon bei annähernder Siedetemperatur aufgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine Masse wird, gegebenenfalls nach Berichtigung, als Prozentsatz der Trockenmasse der Mischung ausgedrückt. Der Anteil an Polychlorid-, Modacryl-, Elasthan, Acetat- und Triacetatfasern in Prozent wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Geräte
3.2. Reagenzien
Anmerkung:
Cyclohexanon ist brennbar und toxisch; beim Gebrauch sind entsprechende Schutzvorkehrungen zu treffen.
4. DURCHFÜHRUNG
Es ist nach den im Allgemeinen Teil angegebenen Anweisungen zu arbeiten und dann folgendermaßen vorzugehen:
100 ml Cyclohexanon je Gramm Probe in den Destillationskolben geben, das Extraktionsgefäß ein- bzw. aufsetzen und den Filtertiegel mit der Probe einführen. Auf den Filtertiegel die leicht geneigte poröse Trennwand legen, danach den Rückflußkühler aufsetzen. Das Cyclohexanon bis zum Siedepunkt erwärmen, 60 Minuten bei einer Mindestgeschwindigkeit von etwa 12 Zyklen extrahieren. Nach Extraktion und Abkühlung den Filtertiegel aus dem Extraktionsgefäß nehmen und die poröse Scheidewand entfernen. Den Inhalt des Filtertiegels 3 bis 4 mal mit auf etwa 60 °C erwärmten 50 prozentigem Äthylalkohol und anschließend mit 1 Liter Wasser bei 60 °C waschen.
Während und zwischen den Waschvorgängen zunächst keinen Unterdruck anwenden, sondern die Flüssigkeit normal ablaufen lassen und erst dann absaugen.
Zum Schluß wird der Tiegel mit dem Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im Allgemeinen Teil angegebenen Berechnungsverfahren berechnet. Der Wert "d" beträgt 1,00, jedoch bei
Seide und Melamin 1,01,
Polyacrylfasern 0,98.
6. GENAUIGKEIT DER ERGEBNISSE
Bei homogenen Textilgemischen liegen die Zuverlässigkeitsgrenzen der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei die statistische Sicherheit 95 % beträgt.

Abbildung gemäß Punkt 3.1 i) des Verfahrens Nr. 15
Verfahren Nr. 16 11
MELAMIN UND BESTIMMTE ANDERE FASERN
(Verfahren mit heißer Ameisensäure)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Mischungen von:
mit
2. GRUNDLAGE DES VERFAHRENS
Die Melaminfasern werden aus einer bekannten Trockenmasse der Mischung mit Hilfe heißer Ameisensäure (90 % Massengehalt) herausgelöst.
Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine Masse wird, gegebenenfalls nach Berichtigung, als Prozentsatz der Trockenmasse der Mischung ausgedrückt. Der Anteil der anderen Bestandteile wird durch Differenzbildung ermittelt.
Anmerkung: Es darf nicht vom empfohlenen Temperaturbereich abgewichen werden, da die Löslichkeit von Melamin in starkem Maße temperaturabhängig ist.
3. GERÄTE UND REAGENZIEN (neben den im Allgemeinen Teil genannten)
3.1. Geräte
3.2. Reagenzien
Beim Umgang mit heißer Ameisensäure ist wegen ihrer stark ätzenden Wirkung äußerste Vorsicht geboten.
4. DURCHFÜHRUNG
Es ist der im Allgemeinen Teil beschriebene Analysevorgang zu befolgen und folgendermaßen vorzugehen:
Die in einem 200-ml-Erlenmeyerkolben mit Schliffstopfen befindliche Probe wird mit 100 ml Ameisensäure je Gramm Probe versetzt. Der Kolben wird verschlossen und geschüttelt, um die Probe vollständig zu benetzen. Der Kolben wird eine Stunde lang in einem Wasserbadschüttler bei 90 ± 2 °C kräftig geschüttelt. Anschließend lässt man den Kolben auf Raumtemperatur abkühlen. Die Flüssigkeit über einen gewogenen Glasfiltertiegel dekantieren. Der Rückstand im Kolben wird mit 50 ml Ameisensäure versetzt und manuell geschüttelt, anschließend wird der Kolbeninhalt durch den Glasfiltertiegel gefiltert. Etwa zurückbleibende Fasern werden durch Auswaschen des Kolbens mit etwas Ameisensäurelösung in den Filtertiegel überführt. Der Filtertiegel wird unter Absaugen entleert und der Rückstand nacheinander mit Ameisensäure, heißem Wasser, verdünnter Ammoniaklösung und schließlich mit kaltem Wasser ausgewaschen, wobei der Tiegel nach jeder Flüssigkeitszugabe unter Absaugen entleert wird, jedoch erst, nachdem die jeweilige Waschlösung durch ihr Eigengewicht abgelaufen ist. Zum Schluss wird der Tiegel unter Absaugen geleert, zusammen mit dem Rückstand getrocknet, abgekühlt und gewogen.
Anmerkung:
Die Löslichkeitseigenschaften von Melamin sind in starkem Maße von der Temperatur abhängig, daher ist diese sorgfältig zu überwachen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im Allgemeinen Teil angegebenen Berechnungsverfahren berechnet. Der Wert "d" für Baumwolle und Aramid beträgt 1,02.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegen die Zuverlässigkeitsgrenzen der Ergebnisse dieses Verfahrens bei höchstens ± 2, wobei die statistische Sicherheit 95 % beträgt.
| Anhang III |
Teil A
Aufgehobene Richtlinien
(nach Artikel 8)
|
| und ihre nachfolgenden Abänderungen: |
|
|
|
Teil B
Umsetzungsfristen
| Richtlinie | Termin für die Umsetzung |
| 72/276/EWG | 18. Januar 1974 |
| 79/76/EWG | 28. Juni 1979 |
| 81/75/EWG | 27. Februar 1982 |
| 87/184/EWG | 1. September 1988 |
| Übereinstimmungstabelle | Anhang IV |
| Diese Richtlinie | Richtlinie 72/276/EWG |
| Artikel 1 | Artikel 1 |
| Artikel 2 | Artikel 2 |
| Artikel 3 | Artikel 3 |
| Artikel 4 | Artikel 4 |
| Artikel 5 | Artikel 5 |
| Artikel 6 | Artikel 6 |
| Artikel 7 | Artikel 7, Absatz 2 |
| Artikel 8 | - |
| Artikel 9 | Artikel 8 |
| Anhang I | Anhang I |
| Anhang II Nummer 1 | Anhang II Nummer 1 |
| Anhang II Nummer 2 | Anhang II Nummer 2 |
| Anhang II Verfahren Nr. 1 | Anhang II Verfahren Nr. 1 |
| Anhang II Verfahren Nr. 2 | Anhang II Verfahren Nr. 2 |
| Anhang II Verfahren Nr. 3 | Anhang II Verfahren Nr. 3 |
| Anhang II Verfahren Nr. 4 | Anhang II Verfahren Nr. 4 |
| Anhang II Verfahren Nr. 5 | Anhang II Verfahren Nr. 5 |
| Anhang II Verfahren Nr. 6 | Anhang II Verfahren Nr. 6 |
| Anhang II Verfahren Nr. 7 | Anhang II Verfahren Nr. 7 |
| Anhang II Verfahren Nr. 8 | Anhang II Verfahren Nr. 8 |
| Anhang II Verfahren Nr. 9 | Anhang II Verfahren Nr. 9 |
| Anhang II Verfahren Nr. 10 | Anhang II Verfahren Nr. 10 |
| Anhang II Verfahren Nr. 11 | Anhang II Verfahren Nr. 11 |
| Anhang II Verfahren Nr. 12 | Anhang II Verfahren Nr. 13 |
| Anhang II Verfahren Nr. 13 | Anhang II Verfahren Nr. 14 |
| Anhang II Verfahren Nr. 14 | Anhang II Verfahren Nr. 15 |
| Anhang II Verfahren Nr. 15 | Anhang II Verfahren Nr. 16 |
| Anhang III | - |
| Anhang IV | - |
 |
ENDE | |
(Stand: 11.03.2019)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: ab 105.- € netto
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion