

umwelt-online: Verordnung (EG) Nr. 429/2008 der Kommission mit Durchführungsbestimmungen zur Verordnung (EG) Nr. 1831/2003 hinsichtlich der Erstellung und Vorlage von Anträgen sowie der Bewertung und Zulassung von Futtermittelzusatzstoffen (2)
|
zurück | |
3.1.2 Untersuchungen in Bezug auf Mikroorganismen
Es werden die Ergebnisse von Untersuchungen vorgelegt, mit denen die Fähigkeit des Zusatzstoffs ermittelt wird, eine Kreuzresistenz gegenüber in der Human- oder Veterinärmedizin verwendeten Antibiotika zu erzeugen, in der Zieltierart unter Feldbedingungen resistente Bakterienstämme zu selektieren, eine Wirkung auf opportunistische Pathogene im Verdauungstrakt hervorzurufen oder ihre Ausscheidung oder die zoonotischer Mikroorganismen zu bewirken.
Besitzen der Wirkstoff bzw. die Wirkstoffe in der mit dem Futtermittel verabreichten Konzentration antimikrobielle Eigenschaften, so wird anhand von Standardverfahren die minimale Hemmkonzentration (MHK) im Hinblick auf relevante Bakterienarten bestimmt. Lässt sich eine einschlägige antimikrobielle Eigenschaft nachweisen, wird die Fähigkeit des Zusatzstoffs bestimmt, in vitro und in der Zieltierart resistente Bakterienstämme zu selektieren, sowie die Fähigkeit, eine Kreuzresistenz gegenüber einschlägigen Antibiotika zu erzeugen 11.
Bei mikrobiellen Zusatzstoffen sowie bei sonstigen Zusatzstoffen, die voraussichtlich Auswirkungen auf die Mikroflora im Darm haben, werden Tests mit der empfohlenen Dosis durchgeführt. Mit diesen Untersuchungen wird belegt, dass die Verwendung des Zusatzstoffs keine Bedingungen schafft, die die übermäßige Vermehrung und die Ausscheidung möglicherweise pathogener Mikroorganismen fördern.
Die Wahl der zu beobachtenden Mikroorganismen hängt von der Zieltierart ab; relevante zoonotische Arten von Mikroorganismen werden einbezogen, unabhängig davon, ob sie bei den Zieltieren Symptome hervorrufen oder nicht.
3.2 Untersuchungen zur Sicherheit der Verwendung des Zusatzstoffs für die Verbraucher
Ziel ist die Beurteilung der Sicherheit des Zusatzstoffs für die Verbraucher und der Nachweis möglicher Rückstände des Zusatzstoffs selbst oder seiner Metaboliten in Lebensmitteln, die aus Tieren gewonnen werden, denen ein mit dem Zusatzstoff versetztes oder behandeltes Futtermittel oder entsprechendes Wasser verabreicht wurde.
3.2.1 Untersuchungen zu Stoffwechsel und Rückständen
Die Ermittlung der Pharmakokinetik des Zusatzstoffs in der Zieltierart ist ein bedeutender Schritt bei der Identifizierung und Quantifizierung der Rückstände in den essbaren Geweben oder essbaren Produkten, die von Tieren gewonnen werden, denen ein den Zusatzstoff enthaltendes Futtermittel oder entsprechendes Wasser verabreicht wurde. Untersuchungen zu Resorption, Verteilung, Stoffwechsel und Ausscheidung der Substanz (und ihrer Metaboliten) müssen vorgelegt werden.
Die Untersuchungen müssen anhand auf internationaler Ebene validierter Testmethoden, in Übereinstimmung mit dem Gemeinschaftsrecht oder mit den OECD-Leitlinien über Methoden und entsprechend den Grundsätzen der GLP durchgeführt werden. Bei den Untersuchungen werden die Gemeinschaftsvorschriften über den Tierschutz eingehalten; die Untersuchungen werden nur wiederholt, wenn dies erforderlich ist.
Bei Untersuchungen zu Stoffwechsel und Rückständen am Zieltier bzw. an den Zieltieren wird der Wirkstoff in das Futtermittel eingebracht (d. h., nicht durch Zwangsfütterung verabreicht, es sei denn, dies wird hinreichend begründet).
Die Struktur der Metaboliten, die mehr als 10 % des Gesamtrückstands in essbaren Geweben und Produkten bzw. mehr als 20 % des Gesamtrückstands in den Exkrementen ausmachen, wird identifiziert. Bestehen hinsichtlich der Pharmakokinetik des Wirkstoffs toxikologische Bedenken, so werden auch Metaboliten mit Werten unterhalb der oben genannten Grenzen identifiziert.
Untersuchungen zur Kinetik der Rückstände bilden die Grundlage für die Berechnung der Exposition der Verbraucher und erforderlichenfalls für die Festlegung von Wartezeit und MRL. Es wird ein Markerrückstand vorgeschlagen.
Für manche Zusatzstoffe können je nach Art und Verwendung Untersuchungen zu Stoffwechsel und Rückständen entfallen.
3.2.1.1 Untersuchungen zum Stoffwechsel
Zweck der Untersuchungen zum Stoffwechsel ist die Beurteilung von Resorption, Verteilung, Biotransformation und Ausscheidung des Zusatzstoffs bei der Zieltierart.
Folgende Untersuchungen werden verlangt:
3.2.1.2 Untersuchungen zu den Rückständen
Gegenstand der Untersuchung sind Menge und Art nicht extrahierbarer Rückstände in essbaren Geweben oder Produkten.
Rückstandsuntersuchungen sind für alle Stoffe erforderlich, für die Untersuchungen zum Stoffwechsel nötig sind.
Ist der Stoff ein natürlicher Bestandteil von Körperflüssigkeiten oder -geweben oder kommt er von Natur aus in großen Mengen in Lebensmitteln oder Futtermitteln vor, beschränkt sich die Notwendigkeit von Rückstanduntersuchungen auf den Vergleich der Mengen in Geweben/Produkten einer Gruppe, welcher der Zusatzstoff nicht verabreicht wurde, und einer Gruppe, der die vorgesehene Höchstdosis verabreicht wurde.
Im Fall von Haupttierarten werden bei der Untersuchung gleichzeitig der toxikologisch relevante Gesamtrückstand bewertet und der Markerrückstand des Wirkstoffs in essbaren Geweben (Leber, Niere, Muskel, Haut, Haut/Fett) und essbaren Produkten (Milch, Eier und Honig) identifiziert. Beim Markerrückstand handelt es sich um einen für die Untersuchung ausgewählten Rückstand, dessen Konzentration in einem bekannten Verhältnis zum toxikologisch relevanten Gesamtrückstand in den Geweben steht. Die Untersuchungen belegen ferner, inwieweit die Rückstände in den Geweben oder Produkten verbleiben, sodass eine angemessene Wartezeit festgelegt werden kann.
Für die Bestimmung der Wartezeit werden folgende Mindestanzahlen von Tieren und/oder Produkten empfohlen, von denen zu jedem Zeitpunkt Proben genommen werden:
Es wird auf eine geeignete Geschlechterverteilung geachtet.
Die Messung der Rückstände erfolgt vor Beginn der Wartezeit (bei Fließgleichgewicht) und zu mindestens drei anderen Zeitpunkten.
Es wird ein Markerrückstand vorgeschlagen.
Untersuchungen zur Resorption, Verteilung und Ausscheidung sowie zur Identifizierung der wichtigsten Metaboliten müssen an der Versuchstierart mit dem niedrigsten NOAEL-Wert (NOAEL: No-Observed-Adverse-Effect-Level) oder ansonsten an Ratten (beide Geschlechter) durchgeführt werden. Möglicherweise sind zusätzliche Untersuchungen zu bestimmten Metaboliten erforderlich, die von der Zieltierart produziert und nicht in nennenswerter Menge von der Versuchstierart gebildet werden.
3.2.1.3 Untersuchungen zu Stoffwechsel und Ausscheidung
Es wird eine Untersuchung zum Stoffwechsel durchgeführt, bei der es um das metabolische Gleichgewicht, das metabolische Profil und die Identifizierung der wichtigsten Metaboliten in Urin und Fäzes geht. Tritt bei einer anderen Versuchstierart ein bedeutender Unterschied zur Sensibilität der Ratte zutage, sind zusätzliche Informationen erforderlich.
3.2.1.4 Bioverfügbarkeit der Rückstände
Bei der Bewertung der Risiken für die Verbraucher aufgrund gebundener Rückstände in tierischen Erzeugnissen kann ein zusätzlicher Sicherheitsfaktor berücksichtigt werden, der auf der Bestimmung der Bioverfügbarkeit der Rückstände unter Einsatz geeigneter Versuchstiere und anerkannter Verfahren beruht.
3.2.2 Toxikologische Untersuchungen
Die Sicherheit des Zusatzstoffs wird auf der Grundlage toxikologischer Untersuchungen bewertet, die in vitro und in vivo an Versuchstieren vorgenommen werden. Hierbei wird im Allgemeinen Folgendes gemessen:
Falls ein Grund zur Besorgnis vorliegt, werden weitere Untersuchungen durchgeführt, die für die Bewertung der Sicherheit des Wirkstoffs und seiner Rückstände nötig sind und zusätzliche Informationen liefern.
Ausgehend von den Ergebnissen dieser Untersuchungen muss für toxikologische Zwecke ein NOAEL-Wert ermittelt werden.
Möglicherweise sind Untersuchungen zu bestimmten Metaboliten erforderlich, die von der Zieltierart produziert und nicht in nennenswerter Menge von der Versuchstierart gebildet werden. Sind die Ergebnisse von Untersuchungen zum Stoffwechsel beim Menschen verfügbar, werden die betreffenden Daten bei der Entscheidung über die Art etwaiger zusätzlicher Untersuchungen in Betracht gezogen.
Gegenstand toxikologischer Untersuchungen muss der Wirkstoff sein. Ist der Wirkstoff in einem Fermentationsprodukt vorhanden, wird dieses untersucht. Das getestete Fermentationsprodukt muss mit demjenigen identisch sein, welches in dem in Verkehr zu bringenden Erzeugnis verwendet werden soll.
Die Untersuchungen müssen anhand auf internationaler Ebene validierter Testmethoden, in Übereinstimmung mit dem Gemeinschaftsrecht oder mit den OECD-Leitlinien über Methoden und entsprechend den Grundsätzen der GLP durchgeführt werden. Bei den Untersuchungen unter Verwendung von Versuchstieren werden die Gemeinschaftsvorschriften über den Tierschutz eingehalten; die Untersuchungen werden nur wiederholt, wenn dies erforderlich ist.
3.2.2.1 Untersuchungen zur akuten Toxizität
Untersuchungen zur akuten Toxizität sind erforderlich, um die Toxizität des Stoffs einzustufen und grob zu beschreiben.
Untersuchungen zur akuten Toxizität werden an mindestens zwei Säugerarten durchgeführt. Gegebenenfalls kann eine Versuchstierart durch die jeweilige Zieltierart ersetzt werden.
Es ist nicht erforderlich, die LD50 exakt zu bestimmen, eine ungefähre Bestimmung der niedrigsten letalen Dosis wird als ausreichend erachtet. Die Höchstdosis soll 2.000 mg pro kg Körpergewicht nicht überschreiten.
Damit die Anzahl und das Leiden der eingesetzten Tiere verringert werden, werden laufend neue Arbeitsvorschriften für Untersuchungen zur akuten Toxizität ausgearbeitet. Nach diesen neuen Arbeitsvorschriften durchgeführte Untersuchungen werden akzeptiert, wenn sie ordnungsgemäß validiert sind.
Die OECD-Leitlinien 402 (Acute Dermal Toxicity), 420 (Acute Oral Toxicity - Fixed Dose Method), 423 (Acute Oral Toxicity - Acute Toxic Class Method) und 425 (Acute Oral Toxicity - Upand-Down Procedure) sollten eingehalten werden.
3.2.2.2 Untersuchungen zur Genotoxizität einschließlich Mutagenität
Um Wirkstoffe und gegebenenfalls ihre Metaboliten und Abbauprodukte zu identifizieren, die mutagene und genotoxische Eigenschaften aufweisen, muss eine ausgewählte Kombination verschiedener Genotoxizitätstests durchgeführt werden. Gegebenenfalls werden die Tests ohne und mit Metabolisierung in Säugetieren durchgeführt; die Eignung des Testsystems für das Testmaterial ist zu berücksichtigen.
Die wichtigsten Tests sind die Folgenden:
Je nach Testergebnis und unter Berücksichtigung des gesamten Toxizitätsprofils des Stoffs sowie seiner vorgesehenen Verwendung sind gegebenenfalls zusätzliche Untersuchungen nötig.
Die Verfahren sollten einer der folgenden OECD-Leitlinien sowie sonstigen einschlägigen OECD-Leitlinien für Invitro- und Invivo-Tests entsprechen:
471 (Salmonella typhimurium Reverse Mutation Test), 472 (Escherichia coli Reverse Mutation Test), 473 (In vitro Mammalian Chromosome Aberration Test), 474 (Mammalian Erythrocyte Micronucleus Test), 475 (Mammalian Bone Marrow Chromosome Aberration Test), 476 (In vitro Mammalian Cell Gene Mutation Test) oder 482 (Genetic Toxicology: DNa Damage and Repair, Unscheduled DNa Synthesis in Mammalian Cells in vitro).
3.2.2.3 Untersuchungen zur subchronischen oralen Toxizität bei wiederholter Verabreichung
Zur Ermittlung der subchronischen Toxizität des Wirkstoffs muss mindestens eine Untersuchung an einer Nagetierart durchgeführt werden, die sich über mindestens 90 Tage erstreckt. Falls dies für erforderlich gehalten wird, muss eine zweite Untersuchung an einer Art vorgenommen werden, die nicht zu den Nagetieren gehört. Hierbei muss der zu testende Wirkstoff in mindestens drei Dosen oral verabreicht werden, und zwar den Versuchstieren und einer Kontrollgruppe, damit eine Dosis-Wirkungs-Beziehung ermittelt werden kann. Normalerweise sollte zu erwarten sein, dass die Höchstdosis schädliche Wirkungen zur Folge hat. Bei der niedrigsten Dosis dürfte nicht zu erwarten sein, dass Anzeichen von Toxizität auftreten.
Die bei diesen Tests genutzten Verfahren sollten der OECD-Leitlinie 408 (Nagetiere) oder 409 (andere Tiere als Nagetiere) entsprechen.
3.2.2.4 Untersuchungen zur chronischen oralen Toxizität (einschließlich Untersuchungen zur Kanzerogenität)
Zur Ermittlung der chronischen Toxizität und der Kanzerogenität muss eine Untersuchung zur chronischen oralen Toxizität an mindestens einer Tierart durchgeführt werden, die sich über mindestens zwölf Monate erstreckt. Eingesetzt wird die Tierart, die auf der Grundlage aller verfügbaren wissenschaftlichen Daten, einschließlich der Ergebnisse der 90-Tage-Studie, am besten geeignet ist. Üblicherweise werden Ratten eingesetzt. Ist eine zweite Untersuchung erforderlich, wird diese an einer Säugerart vorgenommen, die eine Nagetierart sein kann, aber nicht sein muss. Hierbei muss der zu testende Wirkstoff in mindestens drei Dosen oral verabreicht werden, und zwar den Versuchstieren und einer Kontrollgruppe, damit eine Dosis-Wirkungs-Beziehung ermittelt werden kann.
Soll die Untersuchung zur chronischen Toxizität mit einer Untersuchung zur Kanzerogenität gekoppelt werden, verlängert sich die Dauer bei Mäusen und Hamstern auf 18 Monate, bei Ratten auf 24 Monate.
Untersuchungen zur Kanzerogenität können entfallen, wenn der Wirkstoff und seine Metaboliten
Die bei diesen Tests genutzten Verfahren sollten der OECD-Leitlinie
452 (Chronic Toxicity Studies) oder
453 (Combined Chronic Toxicity/Carcinogenicity Studies) entsprechen.
3.2.2.5 Untersuchungen zur Reproduktionstoxizität (einschließlich Untersuchungen zur pränatalen Entwicklungstoxizität)
Zur Ermittlung einer möglichen Beeinträchtigung der männlichen oder weiblichen Fortpflanzungsfähigkeit oder nachteiliger Folgen für die Nachkommenschaft, die sich aus der Verabreichung des Wirkstoffs ergeben, muss die Fortpflanzungsfähigkeit überprüft werden durch
Bei neuartigen Untersuchungen können andere validierte Methoden eingesetzt werden, die den Einsatz von Tieren verringern.
3.2.2.5.1 Untersuchung zur Zwei-Generationen-Reproduktionstoxizität
Es müssen Untersuchungen zur Fortpflanzungsfähigkeit an mindestens einer Tierart, üblicherweise einer Nagetierart, durchgeführt werden, die sich über mindestens zwei Generationen von Nachkommen (F1, F2) erstrecken; sie können mit einer Teratogenitätsstudie kombiniert werden. Der zu prüfende Stoff wird den männlichen und weiblichen Versuchstieren oral zu einem angemessenen Zeitpunkt vor der Paarung verabreicht. Die Verabreichung wird bis zum Absetzen der F2-Generation fortgesetzt.
Fruchtbarkeit, Trächtigkeit, Geburt, Verhalten des Muttertiers, Säugen, Wachstum und Entwicklung der F1-Generation von der Befruchtung bis zur Geschlechtsreife sowie die Entwicklung der F2-Generation bis zum Absetzen müssen sorgfältig beobachtet und dokumentiert werden. Die bei Untersuchungen zur Reproduktionstoxizität genutzten Verfahren sollten der OECD-Leitlinie 416 entsprechen.
3.2.2.5.2 Untersuchung zur pränatalen Entwicklungstoxizität (Untersuchung zur Teratogenität)
Das Ziel besteht darin, etwaige schädliche Wirkungen festzustellen, die sich ab der Implantation während der gesamten Trächtigkeitsdauer aus der Exposition für das trächtige weibliche Tier und die Entwicklung des Embryos und des Fetus ergeben. Zu derartigen Wirkungen gehören eine erhöhte Toxizität beim trächtigen weiblichen Tier, der Tod des Embryo oder des Fetus, ein verändertes Wachstum des Fetus und strukturelle Anomalien sowie Anomalien beim Fetus.
Für die erste Untersuchung sind üblicherweise Ratten die Tierart der Wahl. Ist das Ergebnis hinsichtlich der Teratogenität negativ oder nicht eindeutig, so wird an einer zweiten Tierart, vorzugsweise an Kaninchen, eine weitere Untersuchung zur Entwicklungstoxizität vorgenommen. Ist das Ergebnis der Untersuchung an Ratten hinsichtlich der Teratogenität positiv, so ist eine Untersuchung an einer zweiten Tierart nicht erforderlich, es sei denn, die Prüfung der Ergebnisse sämtlicher wichtiger Untersuchungen ergibt, dass der ADI-Wert auf der Teratogenität bei Ratten beruhen würde. In diesem Fall wäre eine Untersuchung an einer zweiten Tierart nötig, mit der die für diesen Endpunkt empfindlichste Tierart ermittelt wird. Die bei den Tests genutzten Verfahren sollten der OECD-Leitlinie 414 entsprechen.
3.2.2.6 Sonstige besondere toxikologische und pharmakologische Untersuchungen
Falls ein Grund zur Besorgnis vorliegt, werden weitere Untersuchungen durchgeführt, die für die Bewertung der Sicherheit des Wirkstoffs und seiner Rückstände zweckdienlich sind und zusätzliche Informationen liefern. Bei derartigen Untersuchungen können etwa pharmakologische Wirkungen, Wirkungen bei Jungtieren (Tieren vor der Geschlechtsreife), die Immuntoxizität oder Neurotoxizität ermittelt werden.
3.2.2.7 Bestimmung des No-Observed-Effect-Level (NOAEL)
Der NOAEL-Wert beruht im Allgemeinen auf toxikologischen Wirkungen, jedoch sind pharmakologische Wirkungen gegebenenfalls besser als Grundlage geeignet.
Der niedrigste NOAEL-Wert wird ausgewählt. Bei der Ermittlung des niedrigsten NOAEL-Werts, ausgedrückt in mg pro kg Körpergewicht pro Tag, werden sämtliche Ergebnisse aus den vorangegangenen Unterabschnitten und alle sonstigen einschlägigen veröffentlichten Daten (einschließlich etwaiger einschlägiger Informationen über die Wirkung des Wirkstoffs beim Menschen) sowie gegebenenfalls Informationen über chemische Stoffe mit einer sehr ähnlichen chemischen Struktur berücksichtigt.
3.2.3 Bewertung der Sicherheit für die Verbraucher
Die Bewertung der Sicherheit für die Verbraucher erfolgt anhand eines Vergleichs der festgelegten ADI mit der berechneten theoretischen Menge des Zusatzstoffs oder seiner Metaboliten, die die Verbraucher mit Lebensmitteln aufnehmen. Im Fall von Vitaminen und Spurenelementen kann statt des ADI-Werts der UL-Wert (Tolerable Upper Intake Level) verwendet werden.
3.2.3.1 Vorschlag für die tolerierbare tägliche Aufnahme (ADI) des Wirkstoffs bzw. der Wirkstoffe
Die ADI (ausgedrückt in mg des Zusatzstoffs oder der von ihm abgeleiteten Stoffe pro Person und Tag) wird hergeleitet, indem man den niedrigsten NOAEL-Wert durch einen geeigneten Sicherheitsfaktor teilt und mit dem durchschnittlichen menschlichen Körpergewicht von 60 kg multipliziert.
Wenn es erforderlich ist, sollte eine ADI vorgeschlagen werden. Im Fall geringer Toxizität bei Tierversuchen kann in Bezug auf die ADI nicht angegebeneingetragen werden. Eine ADI wird nicht vorgeschlagen, wenn der Stoff Eigenschaften aufweist, die auf den Menschen genotoxisch oder kanzerogen wirken.
Voraussetzung für die Festlegung einer ADI ist, dass die Pharmakokinetik des Wirkstoffs bei den Ziel- und Versuchstieren ähnlich ist (siehe 3.2.1.4Bioverfügbarkeit der Rückstände); dies stellt sicher, dass die Verbraucher gegenüber denselben Rückständen exponiert sind wie die Versuchstiere, die bei toxikologischen Untersuchungen zum Einsatz kommen. Ist keine Ähnlichkeit gegeben, kann mittels zusätzlicher Untersuchungen an einer zweiten Versuchstierart oder anhand der für die Zieltierart typischen Metaboliten trotzdem eine ADI definiert werden.
Bei der Wahl des Sicherheitsfaktors für die Festlegung der ADI eines bestimmten Zusatzstoffs wird Folgendes beachtet: die Art der biologischen Wirkung und die Qualität der Daten, anhand deren der NOAEL-Wert bestimmt wurde; die Bedeutung dieser Wirkung für den Menschen und die Reversibilität der Wirkung; etwaige bekannte unmittelbare Wirkungen der Rückstände beim Menschen.
Bei der Berechnung der ADI wird ein Sicherheitsfaktor von mindestens 100 angewendet (falls eine vollständige Reihe toxikologischer Untersuchungen vorgenommen wurde). Liegen Humandaten zum Wirkstoff vor, so kann ein niedrigerer Sicherheitsfaktor akzeptiert werden. Höhere Sicherheitsfaktoren können angewendet werden, wenn einer zusätzlichen Unsicherheit bei den Daten Rechnung getragen werden soll oder wenn der NOAEL-Wert auf der Grundlage eines besonders entscheidenden Endpunkts, wie etwa der Teratogenität, festgelegt wurde.
3.2.3.2 Zulässige Höchstdosis (Tolerable Upper Intake Level, UL)
Bei manchen Zusatzstoffen ist unter Umständen der UL-Wert besser als Grundlage der Sicherheitsbewertung geeignet; die UL ist die Höchstmenge der gesamten chronischen täglichen Aufnahme eines Nährstoffs (alle Quellen), für den es (laut nationalen oder internationalen wissenschaftlichen Gremien) als unwahrscheinlich gilt, dass er ein Risiko schädlicher Auswirkungen auf die Gesundheit der Verbraucher oder bestimmter Verbrauchergruppen darstellt.
Das Dossier enthält Daten, die belegen, dass die UL, unter Beachtung aller möglichen Quellen des Nährstoffs, durch die Verwendung des Zusatzstoffs nicht überschritten werden kann.
Sind die entsprechenden Rückstandsmengen des ernährungsphysiologischen Zusatzstoffs oder seines Metaboliten bzw. seiner Metaboliten in tierischen Erzeugnissen höher als der bei diesen Erzeugnissen als normal angesehene oder erwartete Wert, ist deutlich darauf hinzuweisen.
3.2.3.3 Exposition der Verbraucher
Die Gesamtmenge des Zusatzstoffs und/oder seiner Metaboliten, die die Verbraucher aus allen Quellen aufnehmen, liegt unter der ADI oder UL.
Die Berechnung der theoretischen Aufnahme durch Lebensmittel tierischen Ursprungs soll unter Berücksichtigung der Konzentration (Gesamtrückstand ausgedrückt durch das arithmetische Mittel und höchster Einzelwert), welche in den Geweben und Produkten nach Anwendungsende gemessen wird, erfolgen. Zusätzlich werden bei Bedarf die Werte für den täglichen menschlichen Verzehr von Lebensmitteln unter Berücksichtigung unterschiedlicher Wartezeiten, ausgehend vom ungünstigsten Fall (worst case scenario) bestimmt.
Bei Zusatzstoffen, die für mehrere Tierarten bestimmt sind, wird die Exposition durch Gewebe für Säugetiere, Vögel und Fische getrennt berechnet und anschließend der höchste Wert genommen. Gegebenenfalls wird diesem Wert die Exposition durch Milch und Eier hinzugerechnet. Wird ein Zusatzstoff etwa Säugetieren in der Laktationsperiode und Legegeflügel verabreicht, werden die jeweiligen Höchstwerte in Bezug auf essbares Gewebe zu denen für den Verzehr von Milch und Eiern dazugezählt. Wird ein Zusatzstoff Fischen, Säugetieren in der Laktationsperiode und Legegeflügel verabreicht, werden die jeweiligen Höchstwerte in Bezug auf essbares Gewebe zu denen für den Verzehr von Milch und Eiern dazugezählt. Andere Kombinationen werden in der gleichen Art und Weise vorgesehen.
Unter bestimmten Bedingungen (z.B. bei ernährungsphysiologischen und sensorischen Zusatzstoffen oder Zusatzstoffen, die für Nebentierarten bestimmt sind) kann es angezeigt sein, in der Folge die Bewertung der Exposition des Menschen anhand realistischerer Zahlen zum Verzehr zu präzisieren, wobei jedoch die konservativste Berechnung beibehalten wird. Wo möglich, stützt sich diese Berechnung auf Daten, die die Europäische Gemeinschaft betreffen.
Tabelle 1 Theoretischer täglicher menschlicher Verzehr (in g Gewebe oder Produkt)
| Säugetiere | Vögel | Fische | Sonstige | |
| Muskel | 300 | 300 | 300 * | |
| Leber | 100 | 100 | - | |
| Niere | 50 | 10 | - | |
| Fett | 50 ** | 90 *** | - | |
| + Milch | 1.500 | - | - | |
| + Eier | - | 100 | - | |
| + Honig | 20 | |||
| *) Muskel und Haut im natürlichen Verhältnis.
**) Beim Schwein: 50 g Fett und Haut im natürlichen Verhältnis. ***) Fett und Haut im natürlichen Verhältnis. |
||||
3.2.3.4 Vorschlag für Rückstandshöchstmengen (Maximum Residue Limits, MRLs)
Die Rückstandshöchstmenge ist die höchste Konzentration von Rückständen (ausgedrückt in µg Markerrückstand pro kg essbares Feuchtgewebe oder Produkt), die von der Gemeinschaft als rechtlich zulässig oder als in Lebensmitteln annehmbar angesehen wird. Sie beruht auf Art und Menge des Rückstands, die laut ADI als für den Menschen toxikologisch ungefährlich eingestuft werden. Liegt kein ADI-Wert vor, so lässt sich eine MRL nicht festlegen.
Bei der Festlegung von MRL für Futtermittelzusatzstoffe werden auch Rückstände aus anderen Quellen (z.B. aus Lebensmitteln pflanzlichen Ursprungs) berücksichtigt. Ferner können die MRL herabgesetzt werden, damit eine Übereinstimmung mit den Verwendungsbedingungen für die jeweiligen Futtermittelzusatzstoffe erreicht wird, und zwar insoweit, als praktische Analysemethoden verfügbar sind.
Gegebenenfalls werden für verschiedene Gewebe der Zieltierart oder für Produkte, die von dieser gewonnen werden, einzelne MRL (ausgedrückt in µg Markerrückstand pro kg essbares Feuchtgewebe oder Produkt) festgelegt. Die einzelnen MRL für verschiedene Gewebe oder Produkte spiegeln die Depletionskinetik sowie die Variabilität der MRL in den betreffenden Geweben/Produkten in Bezug auf die für die Verwendung vorgesehenen Tierarten wider. Die Variabilität wird üblicherweise in Form des Konfidenzintervalls des arithmetischen Mittels (95 %) berücksichtigt. Lässt sich das Konfidenzintervall aufgrund einer geringen Anzahl von Proben nicht berechnen, so wird die Variabilität stattdessen durch den höchsten Einzelwert ausgedrückt.
Untersuchungen zu den MRL für Kokzidiostatika und Histomonostatika müssen entsprechend den einschlägigen geltenden Vorschriften über Tierarzneimittel vorgenommen werden (Notice o to applicants an Guideline, Veterinary medicinal products, Establishment of maximum residue limits (MRLs) for residues of veterinary medicinal products in foodstuffs of animal origin ",, Band 8 der Reihe "The rules governing medicinal products in the European Union ",, Oktober 2005).
Erforderlichenfalls werden Untersuchungen zur Bestimmung von MRL für andere Zusatzstoff-Kategorien als Kokzidiostatika und Histomonostatika gemäß diesem Anhang durchgeführt.
Bei der Bestimmung der Exposition der Verbraucher gegenüber dem Gesamtrückstand (Berechnung gemäß Unterabschnitt 3.2.3.3) wird bei den für die verschiedenen Gewebe oder Produkte vorgesehenen MRL das Verhältnis zwischen Markerrückstand und Gesamtrückstand berücksichtigt (Tabelle 2).
Tabelle 2 Bei der Ableitung einer MRL verwendete Ausdrücke
| i-j | Einzelne Gewebe/Produkte (Leber, Niere, Muskel, Haut + Fett, Milch, Eier, Honig) zu verschiedenen Zeitpunkten |
| MRLi-j | Rückstandshöchstmenge in Geweben/Produkten (mg Marker kg-1) |
| Qti-j | Täglicher menschlicher Verzehr einzelner Gewebe/Produkte (in kg) laut (präzisierten Angaben in) Tabelle 1 |
| TRCi-j | Gesamtrückstandskonzentration in einzelnen Geweben/Produkten (mg kg-1) |
| MRCi-j | Markerrückstandskonzentration in einzelnen Geweben/Produkten (mg kg-1) |
| RMTRi-j | Verhältnis zwischen MRCi-j und TRCi-j in einzelnen Geweben/Produkten |
| DITRi-j | Verzehr bei einzelnen Geweben/Produkten, aus dem Gesamtrückstand errechnet (in mg)
DITRi-j = Qti-j x TRCi-j |
| DITRMRLi-j | Verzehr bei einzelnen Geweben/Produkten, aus den MRL errechnet (in mg)
DITRMRLi-j = Qti-j x MRLi-j x RMTRi-j-1 |
Die gemessenen Werte für TRC und MRC werden gegebenenfalls in die Vorlage in Tabelle 3 eingefügt, die anderen Werte berechnet. Sind die verfügbaren Daten nicht vollständig, weil die Werte unter der Nachweisgrenze (Limit of Detection, LOD) liegen, ist eine Extrapolation des RMTR zulässig.
Eine MRL kann nur dann abgeleitet werden, wenn die Summe der einzelnen DITR-Werte unter der ADI liegt. Wird die ADI überschritten, könnten als Alternative Daten einer längeren Wartezeit oder niedrigerer Dosen genutzt werden. Ein erster Vorschlag für eine MRL lässt sich ableiten, indem man den MRC-Wert als Richtwert heranzieht und die LOQ der Analysemethode berücksichtigt. Die aus den vorgesehenen MRL errechnete Summe der DITRMRL muss unter der ADI und nahe bei der Summe der einzelnen DITR-Werte liegen. Wird die ADI überschritten, wird eine niedrigere MRL vorgeschlagen und der Vergleich wiederholt.
Bei bestimmten Zusatzstoffen könnten Rückstände auftreten, die unterhalb der MRL in Milch, Eiern oder Fleisch liegen, aber die für bestimmte Verfahren der Lebensmittelherstellung erforderliche Lebensmittelqualität beeinträchtigen. Es kann angezeigt sein, für solche Zusatzstoffe zusätzlich zu den MRL einen maximalen mit der Lebensmittelherstellung vereinbaren Rückstand (Maximum (food product) Processing Compatible Residue, MPCR) in Erwägung zu ziehen.
Tabelle 3 Vorlage zur Herleitung des Vorschlags für eine MRL
| Leber | Niere | Muskel | Haut + Fett | Milch | Eier | Honig | Summe | |
| TRC1 (mg kg-1) | - | |||||||
| MRC2 (mg kg-1) | - | |||||||
| RMTR2 | - | |||||||
| DITR3 (mg) | - | |||||||
| Vorgesehene MRL (mg kg-1) | - | |||||||
| DITRMRL (mg) | ||||||||
| 1) Unter Beachtung der vorgesehenen Wartezeit.
2) Idealerweise gleichzeitig mit der TRC bestimmt. 3) Aus TRC-Werten errechnet. |
||||||||
3.2.3.5 Vorschlag für eine Wartezeit
Die Wartezeit umfasst den Zeitraum nach dem Absetzen des Zusatzstoffs, der erforderlich ist, damit die Rückstandsmengen bis unter die MRL sinken können.
3.3 Untersuchungen zur Sicherheit der Verwendung des Zusatzstoffs für Anwender bzw. Arbeitnehmer
Eine Exposition von Arbeitnehmern kann hauptsächlich inhalativ oder topisch bei der Herstellung, Handhabung oder Verwendung des Zusatzstoffs erfolgen. So sind etwa landwirtschaftliche Arbeitnehmer möglicherweise bei der Handhabung oder Mischung des Zusatzstoffs exponiert. Es werden zusätzliche Informationen über die Art und Weise der Handhabung geliefert.
Eine Bewertung des für Arbeitnehmer bestehenden Risikos wird beigefügt. Erfahrungen im Herstellungsbetrieb - so verfügbar - sind häufig eine wichtige Informationsquelle für die Beurteilung des für Arbeitnehmer bestehenden Risikos aufgrund der inhalativen oder topischen Exposition gegenüber dem Zusatzstoff. Besondere Beachtung verdienen Zusatzstoffe bzw. mit Zusatzstoffen behandelte Futtermittel und/oder tierische Exkremente, die in Form trockenen Pulvers vorliegen bzw. eine solche Form annehmen können, sowie Zusatzstoffe mit möglichen allergenem Potenzial.
3.3.1 Bewertung des toxikologischen Risikos für die Sicherheit von Anwendern bzw. Arbeitnehmern
Die Risiken für Arbeitnehmer werden in einer Reihe von Untersuchungen mit dem Zusatzstoff in der Form bewertet, für die der Antrag gestellt wurde. Ist der Zusatzstoff geeignet, einen lungengängigen Staub oder Nebel zu bilden, werden Untersuchungen zur akuten Inhalationstoxizität durchgeführt. Es werden Untersuchungen zur Hautreizung und, im Fall negativer Ergebnisse, zur Reizung der Schleimhäute (z.B. am Auge) vorgenommen. Bewertet wird ferner das allergene Potenzial bzw. die Fähigkeit zur Sensibilisierung der Haut. Die zur Erfüllung der Anforderungen an die Verbrauchersicherheit gewonnenen Toxizitätsdaten (siehe 3.2.2) werden für die Bewertung der potenziellen systemischen Toxizität des Zusatzstoffs herangezogen. Alle diese Daten werden erforderlichenfalls mittels direkter Messung und spezieller Untersuchungen ermittelt.
3.3.1.1 Wirkungen auf das Atmungssystem
Es wird nachgewiesen, dass die aerogenen Mengen an Zusatzstoff-Staub oder -Nebel keine Gesundheitsgefährdung für die Anwender bzw. Arbeitnehmer darstellen. Erforderlichenfalls umfasst dieser Nachweis
Untersuchungen zur akuten Inhalationstoxizität werden vorgenommen, wenn Partikel oder Tröpfchen mit einem Durchmesser von weniger als 50m mehr als ein Gewichtsprozent des Zusatzstoffs ausmachen.
Die bei Untersuchungen zur Inhalationstoxizität genutzten Verfahren sollten der OECD-Leitlinie 403 entsprechen. Werden Untersuchungen zur subchronischen Toxizität als nötig erachtet, so sollte hierbei gemäß den OECD-Leitlinien 412 (Repeated Dose Inhalation Toxicity: 28-day or 14-day Study) oder 413 (Subchronic Inhalation Toxicity: 90-day Study) vorgegangen werden.
3.3.1.2 Wirkungen auf Augen und Haut
Sofern verfügbar, werden direkte Nachweise darüber vorgelegt, dass in bekannten Situationen beim Menschen keine Reizung und/oder Sensibilisierung hervorgerufen wird. In Ergänzung hierzu werden Ergebnisse validierter Tierversuche zur Haut- und Augenreizung sowie zum Sensibilisierungspotenzial in Bezug auf den betreffenden Zusatzstoff eingereicht. Bewertet wird ferner das allergene Potenzial bzw. die Fähigkeit zur Sensibilisierung der Haut. Die bei diesen Untersuchungen genutzten Verfahren sollten den OECD-Leitlinien 404 (Acute Dermal Irritation/Corrosion), 405 (Acute Eye Irritation/Corrosion), 406 (Skin Sensitisation) und 429 (Skin Sensitisation: Local Lymph Node Assay) entsprechen.
Sind ätzende Eigenschaften, entweder aus veröffentlichten Daten oder aufgrund spezieller Invitro-Tests, bekannt, werden keine weiteren Invivo-Tests durchgeführt.
Falls der Zusatzstoff beim Einatmen giftig ist, muss die dermale Toxizität berücksichtigt werden. Die betreffenden Untersuchungen müssen der OECD-Leitlinie 402 (Acute Dermal Toxicity) entsprechen.
3.3.1.3 Systemische Toxizität
Die zur Erfüllung der Anforderungen an die Verbrauchersicherheit sowie sonstiger Anforderungen gewonnenen Toxizitätsdaten (Daten zu Toxizität bei wiederholter Verabreichung, Mutagenität, Kanzerogenität, Fortpflanzungsfähigkeit und Pharmakokinetik) werden für die Bewertung der systemischen Toxizität herangezogen.
3.3.1.4 Expositionsbewertung
Es werden Informationen darüber vorgelegt, auf welche Weise die Verwendung des Zusatzstoffs zur Exposition führen kann (durch Einatmen, über die Haut oder durch orale Aufnahme). Diese Informationen schließen eine quantitative Bewertung ein, sofern eine solche vorliegt, beispielsweise zu der typischen Konzentration in der Luft, zur Hautkontamination oder zur oralen Aufnahme. Liegen keine quantitativen Daten vor, werden ausreichende Informationen geliefert, damit eine angemessene Expositionsbewertung erfolgen kann.
3.3.2 Maßnahmen zur Expositionsbegrenzung
Anhand der Informationen, die die Toxizitäts- und Expositionsbewertung geliefert hat, wird eine Schlussfolgerung bezüglich der Gesundheitsrisiken für Anwender bzw. Arbeitnehmer (Einatmen, Reizung, Sensibilisierung und systemische Toxizität) gezogen. Zur Begrenzung oder Beseitigung der Exposition können Schutzmaßnahmen vorgeschlagen werden. Der Einsatz persönlicher Schutzausrüstungen wird allerdings nur als letzter Ausweg gesehen, und zwar zum Schutz vor etwaigen Restrisiken, die nach Einführung von Schutzmaßnahmen verbleiben. So ist es etwa zweckmäßiger, die Neuformulierung des Produkts in Betracht zu ziehen.
3.4 Untersuchungen zur Umweltsicherheit bei der Verwendung des Zusatzstoffs
Es ist wichtig, die Auswirkungen von Zusatzstoffen auf die Umwelt zu betrachten, da diese typischerweise über einen langen Zeitraum hinweg verabreicht werden, oft große Gruppen von Tieren betroffen sind und der Wirkstoff bzw. die Wirkstoffe großteils in Form der Muttersubstanz oder ihrer Metaboliten ausgeschieden werden.
Bei der Bestimmung der Auswirkungen von Zusatzstoffen auf die Umwelt wird schrittweise vorgegangen. In Phase I müssen alle Zusatzstoffe bewertet werden, damit sich diejenigen Zusatzstoffe identifizieren lassen, die keiner weiteren Prüfung bedürfen. Für die übrigen Zusatzstoffe ist eine zweite Phase der Bewertung (Phase II) erforderlich, und zwar zwecks Gewinnung zusätzlicher Informationen, auf deren Grundlage sich weitere Untersuchungen als notwendig erweisen können. Diese Untersuchungen werden gemäß der Richtlinie 67/548/ EWG durchgeführt.
3.4.1 Phase-I-Bewertung
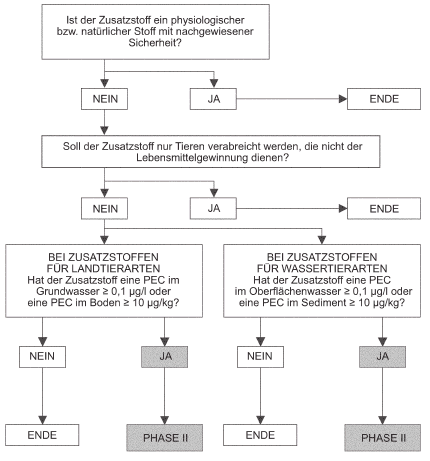
Zweck der Phase-I-Bewertung ist es zu beurteilen, ob eine signifikante Auswirkung des Zusatzstoffs oder seiner Metaboliten wahrscheinlich und eine Phase-II-Bewertung erforderlich ist (siehe den Entscheidungsbaum).
Auf Phase II kann in zwei Fällen verzichtet werden, es sei denn, es liegen wissenschaftliche Hinweise vor, die Grund zu Bedenken geben:
Kann der Antragsteller nicht nachweisen, dass auf den Zusatzstoff eine dieser Ausnahmen zutrifft, so ist eine Phase-II-Bewertung erforderlich.
3.4.1.1 Für Landtiere bestimmte Zusatzstoffe
Werden Exkremente von Nutztieren auf dem Acker ausgebracht, kann die Verwendung von Futtermittelzusatzstoffen zur Kontamination von Boden, Grundwasser und Oberflächenwasser (aufgrund von Drainage und Abschwemmung) führen.
Die im schlimmsten Fall zu erwartende PEC im Boden (PECsoil) würde dadurch entstehen, dass alle ausgeschiedenen Verbindungen auf dem Acker ausgestreut werden. Beträgt die PECsoil (Standardtiefe: 5 cm) weniger als 10 µg/kg, ist keine weitere Bewertung erforderlich.
Weist die PEC für die Kontamination von Grundwasser (PECgw) weniger als 0,1 µg/l auf, ist keine Phase-IIBewertung der ökologischen Auswirkungen des Zusatzstoffs auf das Grundwasser erforderlich.
3.4.1.2 Für Wassertiere bestimmte Zusatzstoffe
In Aquakulturen verwendete Futtermittelzusatzstoffe können zu einer Kontamination von Sedimenten und Wasser führen. Bei in Käfigen gezüchteten Fischen wird das Sediment als das Medium angenommen, das für die Bewertung der Umweltrisiken von Belang ist. Bei an Land gezüchteten Fischen wird angenommen, dass das Abwasser, das zum Oberflächenwasser fließt, das größte Umweltrisiko darstellt.
Die im schlimmsten Fall zu erwartende PEC in den Sedimenten (PECsediment) würde dadurch entstehen, dass sich alle ausgeschiedenen Verbindungen in den Sedimenten ablagern. Beträgt die PECsediment (Standardtiefe: 20 cm) weniger als 10 µg/kg Nassgewicht, ist keine weitere Bewertung erforderlich.
Weist die PEC im Oberflächenwasser (PECsw) weniger als 0,1 µg/l auf, ist keine weitere Bewertung erforderlich.
Entscheidungsbaum für Phase I
3.4.2 Phase-II-Bewertung
Zweck der Phase II ist es, das Potenzial von Zusatzstoffen zu bewerten, Tierarten in der Umwelt, die nicht zu den Zieltierarten gehören, einschließlich Wasser- und Landtieren, zu schädigen oder in unannehmbaren Mengen in das Grundwasser zu gelangen. Aus praktischer Sicht ist es nicht sinnvoll, die Auswirkungen eines Zusatzstoffs auf jede Tierart in der Umwelt zu bewerten, die gegenüber dem Zusatzstoff exponiert sein kann, nachdem dieser den Zieltierarten verabreicht wurde. Die untersuchten taxonomischen Einheiten sollen als Ersatz oder Indikator für das Spektrum der in der Umwelt vorhandenen Tierarten dienen.
Die Phase-II-Bewertung beruht auf einem Risikoquotienten, der für jedes Medium mittels Vergleich der berechneten Werte der PEC und der vorausgesagten Konzentration, bei der keine Wirkung auftritt (Predicted No Effect Concentration, PNEC), ermittelt wird. Die PNEC wird berechnet, indem in Versuchen bestimmte Endpunkte durch einen geeigneten Bewertungsfaktor dividiert werden. Der PNEC-Wert wird für jedes Medium gesondert berechnet.
Die Phase-II-Bewertung beginnt mit einer genaueren Bestimmung der PEC, falls dies möglich ist, und bedient sich zur Bewertung der Umweltrisiken eines zweistufigen Verfahrens.
Die erste Stufe, Phase IIA, besteht aus einer begrenzten Anzahl von Untersuchungen zur Pharmakokinetik und zu den Auswirkungen, die eine konservative Bewertung des Risikos auf der Grundlage der Exposition und der Auswirkungen im betrachteten Umweltmedium ermöglichen soll. Ist der PEC-PNEC-Quotient niedriger als eins (1), ist keine weitere Bewertung erforderlich, es sei denn, eine Bioakkumulation wird erwartet.
Weist der PEC-PNEC-Quotient auf ein unannehmbares Risiko (Quotient > 1) hin, geht der Antragsteller zur Phase IIB über, um die Umweltrisiken genauer zu bewerten.
3.4.2.1 Phase IIA
Zusätzlich zu den in Phase I betrachteten Medien muss auch für Oberflächenwasser die PEC berechnet werden; dabei werden Abschwemmung und Drainage berücksichtigt.
Auf der Grundlage von Daten, die in Phase I nicht herangezogen wurden, lässt sich für jedes betrachtete Umweltmedium ein genauerer PEC-Wert errechnen. Dabei werden folgende Punkte berücksichtigt:
Für die Zwecke der Risikobewertung in Phase II wird für jedes betrachtete Umweltmedium der höchste auf diese Weise errechnete PEC-Wert herangezogen.
Ist mit einer hohen Persistenz im Boden bzw. in den Sedimenten zu rechnen (Zeit für den Abbau von 90 % der Verbindung in ihrer ursprünglichen Konzentration (DT90) > 1 Jahr), wird das Akkumulationspotenzial berücksichtigt.
Es werden diejenigen Mengen an Zusatzstoffen (oder Metaboliten) bestimmt, die auf unterschiedlichen trophischen Ebenen in den betrachteten Umweltmedien ernste schädliche Wirkungen hervorrufen. Es handelt sich hierbei meistens um Untersuchungen zur akuten Toxizität, die den OECD-Leitlinien oder Leitlinien mit vergleichbarem Status entsprechen sollten. Die Untersuchungen in Bezug auf den Boden umfassen Regenwürmer (Toxizität), drei Bodenpflanzen und im Boden lebende Mikroorganismen (z.B. Auswirkungen auf die Stickstoff-Fixierung). Die Untersuchungen für das Süßwassermilieu umfassen Fische (Toxizität), Daphnia magna, Algen und einen im Sediment lebenden Organismus. Im Fall von Meereskäfigen werden drei Arten im Sediment lebender Organismen aus verschiedenen Taxa untersucht.
Für jedes zu betrachtende Medium wird der PNEC-Wert berechnet. In der Regel ergibt sich der PNEC-Wert, indem der niedrigste in den genannten Untersuchungen beobachtete Toxizitätswert durch einen Sicherheitsfaktor von mindestens 100 dividiert wird, der vom Endpunkt und der Zahl der eingesetzten Versuchstierarten abhängt.
Das Bioakkumulationspotenzial kann ausgehend vom Wert des Verteilungskoeffizienten n-Oktanol/Wasser (Log Kow) geschätzt werden. Werte ≥3 weisen daraufhin, dass der Stoff möglicherweise bioakkumuliert wird. Was die Bewertung des Risikos einer Sekundärvergiftung betrifft, so wird ausgelotet, ob eine Untersuchung zur Ermittlung des Biokonzentrationsfaktors (BCF) in Phase IIB durchgeführt werden soll.
3.4.2.2 Phase IIB (eingehendere toxikologische Untersuchungen)
Lässt sich bei einem Zusatzstoff nach der Phase-IIA-Bewertung ein Umweltrisiko nicht ausschließen, so sind genauere Informationen über die Auswirkungen auf die Arten in dem Umweltmedium bzw. in den Umweltmedien erforderlich, in denen die Phase-IIA-Untersuchungen mögliche Probleme erkennen lassen. In diesem Fall sind weitere Untersuchungen nötig, bei denen die chronischen und spezifischeren Auswirkungen auf entsprechende Mikroben-, Pflanzen- und Tierarten bestimmt werden. Die so gewonnenen zusätzlichen Informationen erlauben es, einen niedrigeren Sicherheitsfaktor anzuwenden.
Geeignete zusätzliche Ökotoxizitätstests werden in einer Reihe von Veröffentlichungen erläutert, unter anderem in den OECD-Leitlinien. Die Tests sind sorgfältig auszuwählen, damit sichergestellt wird, dass sie sich für die Bedingungen eignen, unter denen der Zusatzstoff und/oder seine Metaboliten in die Umwelt freigesetzt und dort verbreitet werden. Die genauere Bewertung der Auswirkungen auf den Boden (PNECsoil) könnte sich auf Folgendes stützen: Untersuchungen zu den chronischen Auswirkungen auf Regenwürmer, zusätzliche Untersuchungen zur Bodenmikroflora und zu einigen einschlägigen Pflanzenarten sowie Untersuchungen an Wirbellosen des Grünlands (etwa Insekten) und an Wildvögeln.
Eine genauere Bewertung der Auswirkungen auf Wasser bzw. Sedimente könnte sich auf Untersuchungen zur chronischen Toxizität für die empfindlichsten aquatischen bzw. benthonischen Organismen stützen, die im Zuge der Phase-IIA-Bewertung identifiziert wurden.
Allenfalls erforderliche Untersuchungen zur Bioakkumulation sollten gemäß der OECD-Leitlinie 305 durchgeführt werden.
4. Abschnitt IV: Untersuchungen zur Wirksamkeit des Zusatzstoffs
Durch Untersuchungen wird die Wirksamkeit für jede vorgesehene Verwendung belegt und nachgewiesen, dass der Zusatzstoff zumindest eine der Eigenschaften gemäß Artikel 5 Absatz 3 der Verordnung (EG) Nr. 1831/ 2003 aufweist und hierbei den Kategorien und Funktionsgruppen von Futtermittelzusatzstoffen gemäß Artikel 6 und Anhang I der genannten Verordnung entspricht. Außerdem müssen derartige Untersuchungen die Beurteilung der Wirksamkeit des Zusatzstoffs entsprechend der gemeinsamen landwirtschaftlichen Praxis in der EU ermöglichen.
Die Versuchskonzeption muss entsprechend der Verwendung des Zusatzstoffs, der Tierart und -kategorie begründet werden. Untersuchungen mit Versuchstieren werden so durchgeführt, dass deren Gesundheit und deren Haltungsbedingungen die Beurteilung der Ergebnisse nicht beeinflussen. Für jeden Versuch werden die positiven und negativen Wirkungen technologischer und biologischer Art beschrieben. Auch wird nachgewiesen, dass keine Wirkungen vorliegen, die die Beschaffenheit der tierischen Erzeugnisse beeinträchtigen. Im Idealfall entsprechen die Untersuchungen den Kriterien eines anerkannten, extern auditierten Qualitätssicherungssystems. Ist ein solches System nicht vorhanden, wird nachgewiesen, dass qualifiziertes Personal unter Aufsicht eines namentlich genannten Prüfleiters die Arbeiten durchgeführt und zu diesem Zweck geeignete Einrichtungen und Geräte genutzt hat.
Das Versuchsprotokoll wird vom Prüfleiter sorgfältig angefertigt und enthält z.B. Angaben zu den eingesetzten Methoden, Geräten und Materialien, sowie genaue Informationen über Art, Rasse oder Stamm der Tiere, über deren Anzahl und über die Bedingungen, unter denen sie gehalten und gefüttert wurden. Bei allen Untersuchungen mit Tieren werden die Versuchsbedingungen gemäß 3.1.1.3 erläutert. Endberichte, Rohdaten, Prüfpläne und genau beschriebene und eindeutig identifizierte Testsubstanzen werden archiviert, damit diese später als Referenz zur Verfügung stehen können.
Die Untersuchungen sollen so gestaltet werden, dass sie die Wirksamkeit bei der empfohlenen Mindestdosis des Zusatzstoffs in Bezug auf sensible Parameter im Vergleich zu einer negativen, und falls gewünscht, zu einer positiven Kontrollgruppe nachweisen. Bei derartigen Untersuchungen wird, falls vorgeschlagen, auch die empfohlene Höchstdosis verabreicht. Damit wissenschaftliche Flexibilität und Spielräume bei der Planung und Durchführung der Untersuchungen möglich sind, wird kein bestimmtes Design empfohlen.
Darüber hinaus sind bekannte oder potenzielle biologische oder chemische Wechselwirkungen zwischen dem Zusatzstoff, anderen Zusatzstoffen und/oder Tierarzneimitteln und/oder Bestandteilen der Ration besonders zu beachten, sofern dies für die Wirksamkeit des betreffenden Zusatzstoffs von Bedeutung ist (z.B. Kompatibilität eines mikrobiellen Zusatzstoffs mit Kokzidiostatika und Histomonostatika oder mit organischen Säuren).
4.1 Invitro-Untersuchungen
Bei allen technologischen und bestimmten sensorischen Zusatzstoffen, die die Eigenschaften von Futtermitteln beeinflussen, wird die Wirksamkeit mittels einer Laboruntersuchung nachgewiesen. Die Untersuchung deckt ein repräsentatives Spektrum von Futtermitteln ab, bei denen der Zusatzstoff angewendet wird. Die Ergebnisse werden vorzugsweise anhand parameterfreier Tests beurteilt und sichern die erwarteten Änderungen mit einer Wahrscheinlichkeit von p ≤ 0,05.
Invitro-Untersuchungen, insbesondere solche, bei denen Wirkungsweisen des Verdauungstrakts simuliert werden, können zum Nachweis der Wirksamkeit bei anderen Arten von Zusatzstoffen genutzt werden. Diese Untersuchungen sollten eine statistische Auswertung erlauben.
4.2 Kurzzeit-Wirksamkeitsstudien an Tieren
Zum Zweck des Nachweises, inwieweit ein bereits zugelassener oder bekannter gleichwertiger Zusatzstoff durch eine neue Form oder Herkunft eines Nährstoffs oder Farbstoffs ersetzbar ist, können Untersuchungen zur Bioverfügbarkeit herangezogen werden.
Durch Verdaulichkeits- bzw. Bilanzstudien können Untersuchungen zur Leistung der Tiere unterstützt und so die Wirkungsweise belegt werden. In manchen Fällen, insbesondere in Bezug auf eine positive Beeinflussung der Umwelt, kann die Wirksamkeit besser durch Bilanzversuche nachgewiesen und, diese können Langzeit-Wirksamkeitsstudien vorgezogen werden. Bei derartigen Versuchen richten sich Anzahl und Art bzw. Kategorie der Tiere nach den vorgesehenen Verwendungsbedingungen.
Gegebenenfalls können andere Kurzzeit-Wirksamkeitsstudien an Tieren vorgeschlagen werden, durch die sich entsprechende Langzeit-Wirksamkeitsstudien ersetzen lassen, wenn dies umfassend begründet wird.
4.3 Langzeit-Wirksamkeitsstudien an Tieren
Die Untersuchungen sollten an mindestens zwei verschiedenen Orten durchgeführt werden.
Bei der Wahl der Versuchskonzeption muss für eine ausreichende statistische Aussagekraft gesorgt werden; Risiken des Typs 1 und 2 müssen berücksichtigt werden. Das Verfahren muss ausreichend empfindlich sein, damit jede Wirkung des Zusatzstoffs bei Verabreichung der empfohlenen Mindestdosis ermittelt wird (Typ-1-α-Risiko, p ≤ 0,05 im Allgemeinen und p ≤ 0,1 im Fall von Wiederkäuern, Nebentierarten, Heimtieren und nicht der Lebensmittelgewinnung dienenden Tieren), sowie über eine ausreichende statistische Aussagekraft verfügen; dadurch soll gewährleistet werden, dass mit der Analysemethode das Ziel der Untersuchung erreicht wird. Das Typ-2-β-Risiko weist im Allgemeinen einen Wert von höchstens 20 % auf, bei Versuchen mit Wiederkäuern, Nebentierarten, Heimtieren und nicht der Lebensmittelgewinnung dienenden Tieren beträgt es höchstens 25 %; daher hat das Risiko eine statistische Aussagekraft (1β) von mindestens 80 % (bei Wiederkäuern, Nebentierarten, Heimtieren und nicht der Lebensmittelgewinnung dienenden Tieren 75 %).
Es ist bekannt, dass die Eigenschaften mancher Zusatzstoffe die Festlegung von Versuchsbedingungen erschweren, unter denen sich optimale Ergebnisse erzielen lassen. Daher wird die Möglichkeit einer Metaanalyse in Betracht gezogen, wenn die Ergebnisse von mehr als drei Untersuchungen vorliegen. Aus diesem Grunde kommen bei sämtlichen Untersuchungen die gleichen Verfahren zum Einsatz, sodass abschließend die Daten auf ihre Homogenität getestet und (bei positivem Testergebnis) für die statistische Auswertung bei p ≤0,05 zusammengefasst werden können.
4.4 Dauer von Langzeit-Wirksamkeitsstudien an Tieren
In der Regel entspricht die Dauer von Wirksamkeitsstudien dem im Antrag angeführten Zeitraum.
Wirksamkeitsstudien werden gemäß der landwirtschaftlichen Praxis in der Europäischen Union vorgenommen, ihre Mindestdauer ist in Anhang IV festgelegt.
Wird ein Futtermittel für eine bestimmte Dauer verabreicht, die kürzer ist als die, die sich aus der Definition der Tierkategorie ergibt, erfolgt dies entsprechend den vorgesehenen Verwendungsbedingungen. Allerdings ist der Beobachtungszeitraum nicht kürzer als 28 Tage, und die einschlägigen Endpunkte (z.B. bei Zuchtsauen die Anzahl der lebend geborenen Ferkel unter Beachtung der Trächtigkeitsdauer, oder die Anzahl und das Gewicht der abgesetzten Ferkel unter Berücksichtigung der Laktationsperiode) werden in die Beobachtung einbezogen.
Bei anderen Tierarten oder -kategorien, für die in Anhang IV in Bezug auf Wirksamkeitsstudien keine Mindestdauer festgelegt ist, wird eine Verabreichungsdauer entsprechend den vorgesehenen Verwendungsbedingungen in Betracht gezogen.
4.5 Anforderungen an die Wirksamkeit von Zusatzstoffen der einzelnen Kategorien und Funktionsgruppen
Bei allen Zusatzstoffen, die eine Wirkung auf Tiere haben sollen, werden Invivo-Wirksamkeitsstudien verlangt.
Bei zootechnischen Zusatzstoffen, Kokzidiostatika und Histomonostatika wird die Wirksamkeit durch mindestens drei Langzeit-Wirksamkeitsstudien nachgewiesen. Bei bestimmten zootechnischen Zusatzstoffen und Zusatzstoffen anderer Kategorien, die eine Wirkung auf Tiere haben, können Kurzzeit-Wirksamkeitsstudien akzeptiert werden, wenn sich die Wirksamkeit eindeutig nachweisen lässt.
Bei den übrigen Kategorien von Zusatzstoffen, die keine unmittelbare Wirkung auf Tiere haben, wird mindestens eine Invitro-Wirksamkeitsstudie durchgeführt.
4.6 Untersuchungen zur Qualität tierischer Erzeugnisse, wenn Auswirkungen auf diese nicht Gegenstand des Antrags sind
Zum Nachweis, dass der Zusatzstoff keine negative oder andere unerwünschte Wirkung auf die organoleptischen und ernährungsphysiologischen (gegebenenfalls hygienischen und technologischen) Eigenschaften von Lebensmitteln hat, die von Tieren gewonnen werden, denen der Zusatzstoff verfüttert wurde, werden bei einer der Wirksamkeitsstudien geeignete Stichproben vorgenommen. Zwei Gruppen werden beobachtet: eine Gruppe, der der Zusatzstoff nicht verabreicht wurde; eine Gruppe, der die vorgesehene Höchstdosis verabreicht wurde. Die Daten ermöglichen eine statistische Auswertung. Ein Fehlen dieser Untersuchungen ist entsprechend zu begründen.
5. Abschnitt V: Plan zur Marktbegleitenden Beobachtung
Gemäß Artikel 7 Absatz 3 Buchstabe g der Verordnung (EG) Nr. 1831/2003 und entsprechend den Eigenschaften der betreffenden Zusatzstoffe wird bei bestimmten Kategorien von Zusatzstoffen ein Vorschlag für die marktbegleitende Beobachtung vorgelegt, damit alle direkten oder indirekten, unmittelbaren oder späteren sowie unvorhergesehenen Auswirkungen der Verwendung des Zusatzstoffs auf die Gesundheit von Mensch oder Tier oder auf die Umwelt verfolgt und festgestellt werden können.
Der Beobachtungsplan wird von Fall zu Fall gestaltet und dieser regelt, wer (z.B. Antragsteller, Anwender) die gemäß diesem Plan erforderlichen Aufgaben wahrnimmt, wer sicherstellen muss, dass der Beobachtungsplan ordnungsgemäß aufgestellt und umgesetzt wird und wer gewährleistet, dass ein Verfahren besteht, nach dem die zuständigen Behörden etwaige neue Informationen zur Sicherheit der Verwendung des Zusatzstoffs erhalten. Unbeschadet der Bestimmungen über die Überwachung gemäß Artikel 12 der Verordnung (EG) Nr. 1831/2003 werden die Kommission und die Behörde über jede beobachtete negative Auswirkung unterrichtet.
Ist der Wirkstoff gleichzeitig ein bekanntes Antibiotikum und ist er nachweislich in der Lage, bei Verwendung in Futtermitteln resistente Bakterienstämme zu selektieren, so sind im Rahmen der marktbegleitenden Beobachtung Feldversuche durchzuführen, um die bakterielle Resistenz gegen den Zusatzstoff zu überwachen.
Bei Kokzidiostatika und Histomonostatika erfolgt eine Überwachung im Feld auf die Resistenz von Eimeria spp. bzw. Histomonas meleagridis, vorzugsweise während der letzen Phase der Geltungsdauer der Zulassung.
________________
1) ABl. L 50 vom 20.02.2004 S. 44.
2) ABl. L 196 vom 16.08.1967 S. 1. Richtlinie zuletzt geändert durch die Richtlinie 2006/121/EG des Europäischen Parlaments und des Rates (ABl. L 396 vom 30.12.2006 S. 852). Berichtigte Fassung im ABl. L 136 vom 29.05.2007 S. 281).
3) ABl. L 152 vom 30.04.2004 S. 1. Berichtigte Fassung im ABl. L 216 vom 16.06.2004 S. 3.
4) ABl. L 165 vom 30.04.2004. Berichtigte Fassung im ABl. L 191 vom 28.05.2004 S. 1.
5) ABl. L 117 vom 08.05.1990 S. 1. Richtlinie zuletzt geändert durch Entscheidung 2005/174/EG der Kommission (ABl. L 59 vom 05.03.2005 S. 20).
6) ABl. L 76 vom 22.03.1991 S. 35. Richtlinie zuletzt geändert durch die Richtlinie 2001/58/EG (ABl. L 212 vom 07.08.2001 S. 24).
7) ABl. L 262 vom 17.10.2000 S. 21.
8) ABl. L 224 vom 18.08.1990 S. 1. Verordnung zuletzt geändert durch die Verordnung (EG) Nr. 203/2008 (ABl. L 60 vom 05.03.2008 S. 18).
9) M. Thompson et al., Harmonized Guidelines For Single-Laboratory Validation Of Methods Of Analysis (Harmonisierter Leitfaden für die Validierung von Analysenmethoden durch Einzellaboratorien) (IUPAC Technical Report), Pure and Applied Chemistry, Vol. 74, No. 5, S. 835-855, 2002.
10) ABl. L 221 vom 17.08.2002 S. 8. Entscheidung zuletzt geändert durch die Entscheidung 2004/25/EG (ABl. L 6 vom 10.01.2004 S. 38).
11) Eine nicht abschließende Liste ist verfügbar unter www.efsa.europa.eu/en/science/feedap/feedap_opinion/993.html.
| Besondere Anforderungen an das Dossier gemäss Artikel 3 hinsichtlich bestimmter Kategorien von Zusatzstoffen oder im Hinblick auf besondere Fälle gemäss Artikel 7 Absatz 5 der Verordnung (EG) Nr. 1831/2003 | Anhang III |
Die Verordnung (EG) Nr. 1831/2003 sieht erforderlichenfalls für jede einzelne Kategorie von Zusatzstoffen oder für sonstige besondere Ziele gemäß Artikel 7 Absatz 5 der Verordnung (EG) Nr. 1831/2003 eine zusätzliche Unterstützung bei der Erstellung von Dossiers vor.
Verzeichnis der besonderen Anforderungen an die Erstellung von Dossiers für
Fehlen im Dossier Daten, die in diesen Abschnitten vorgesehen sind, so ist dies zu begründen.
1. Technologische Zusatzstoffe
1.1 Abschnitt I: Zusammenfassung des Dossiers
Anhang II Abschnitt I kommt uneingeschränkt zur Anwendung.
1.2 Abschnitt II: Identifizierung, Merkmale und Anwendungsbedingungen des Zusatzstoffs sowie 1.2. Analysemethoden
Anhang II Abschnitt II kommt uneingeschränkt zur Anwendung.
1.3 Abschnitt III: Untersuchungen zur Sicherheit des Zusatzstoffs
Anhang II Unterabschnitte 3.1, 3.2 und 3.4 finden nicht auf Silierzusatzstoffe Anwendung, für die sich nachweisen lässt, dass
In allen anderen Fällen gilt Anhang II Abschnitt III uneingeschränkt.
1.3.1 Untersuchungen zur Anwendungssicherheit des Zusatzstoffs bei den Zieltierarten
Für Xenobiotika 1 gilt Anhang II Unterabschnitt 3.1 uneingeschränkt. 1.3.1.1. Untersuchungen zur Toleranz bei den Zieltierarten
Silierzusatzstoffe:
Toleranztests lassen sich normalerweise auf eine Art von Wiederkäuer, in der Regel Milchkühe, begrenzen. Untersuchungen an anderen Tierarten sind nur dann erforderlich, wenn das silierte Material aufgrund seiner Eigenschaften für die Verwendung bei Tierarten, die keine Wiederkäuer sind, besser geeignet ist.
Andere Stoffe:
Bei anderen Stoffen, die laut Antrag als technologische Zusatzstoffe zugelassen werden sollen und noch nicht für die Verwendung in Futtermitteln zugelassen sind, wird nachgewiesen, dass die vorgeschlagene Höchstdosis die Tiere nicht schädigt. Dieser Nachweis kann sich auf eine Untersuchung an einer der empfindlichsten Zieltierarten oder an einer Versuchstierart beschränken.
1.3.1.2 Untersuchungen in Bezug auf Mikroorganismen
Anhang II Unterabschnitt 3.1.2 kommt uneingeschränkt zur Anwendung.
1.3.2 Untersuchungen zur Sicherheit der Verwendung des Zusatzstoffs für Verbraucher
1.3.2.1 Untersuchungen zu Stoffwechsel und Rückständen
Untersuchungen zu Stoffwechsel und Rückständen sind nicht erforderlich, wenn
Auch sind Untersuchungen zum Stoffwechsel nicht erforderlich, wenn der Stoff von Natur aus in großen Mengen in Lebensmitteln oder Futtermitteln vorkommt oder ein natürlicher Bestandteil von Körperflüssigkeiten oder -geweben ist. In den Fällen, in den Untersuchungen zu den Rückständen verlangt werden, können sich diese jedoch auf den Vergleich der Mengen in Geweben/Produkten bei einer Gruppe, der der Zusatzstoff nicht verabreicht wurde, und bei einer Gruppe, der die empfohlene Höchstdosis verabreicht wurde, beschränken.
1.3.2.2 Toxikologische Untersuchungen
Toxikologische Untersuchungen sind nicht erforderlich, wenn
Bei Mikroorganismen und Enzymen, auf die nicht eine der oben genannten Ausnahmen zutrifft, sind Untersuchungen zur Genotoxizität (einschließlich Mutagenität) und eine Untersuchung zur subchronischen oralen Toxizität nötig. Untersuchungen zur Genotoxizität werden nicht durchgeführt, wenn lebende Zellen vorhanden sind.
Für Xenobiotika, auf die nicht eine der oben genannten Ausnahmen zutrifft, gilt Anhang II Unterabschnitt 3.2.2 uneingeschränkt.
Bei anderen Stoffen wird von Fall zu Fall über die Vorgehensweise entschieden; hierbei werden Grad und Pfade der Exposition berücksichtigt.
1.3.2.3 Bewertung der Sicherheit für die Verbraucher
Anhang II Unterabschnitt 3.2.3 findet uneingeschränkt auf Zusatzstoffe Anwendung, deren Zulassung für Tiere beantragt wird, die der Lebensmittelgewinnung dienen.
1.3.3 Untersuchungen zur Sicherheit der Verwendung des Zusatzstoffs für Anwender bzw. Arbeitnehmer
Anhang II Unterabschnitt 3.3 kommt uneingeschränkt zur Anwendung. Zusatzstoffe, die Enzyme und Mikroorganismen enthalten, gelten als das Atmungssystem sensibilisierend, es sei denn, ein überzeugender Nachweis wird erbracht, der das Gegenteil belegt.
1.3.4 Untersuchungen zur Umweltsicherheit der Verwendung des Zusatzstoffs
Anhang II Unterabschnitt 3.4 kommt uneingeschränkt zur Anwendung. Bei Silierzusatzstoffen werden die Auswirkungen des Zusatzstoffs auf das Entstehen von Sickersaft beachtet, der während des Silierens aus der Miete oder dem Silo fließen.
1.4 Abschnitt IV: Untersuchungen zur Wirksamkeit des Zusatzstoffs
Technologische Zusatzstoffe sollen die Eigenschaften von Futtermitteln verbessern oder stabilisieren, haben aber im Allgemeinen keine direkten biologischen Auswirkungen auf die Tierproduktion. Die Wirksamkeit des Zusatzstoffs muss anhand geeigneter Kriterien, die in anerkannten Verfahren herangezogen werden, durch einen Vergleich mit geeigneten Kontrollfuttermitteln unter den für die Praxis vorgesehenen Verwendungsbedingungen nachgewiesen werden.
Die Wirksamkeit wird anhand von Invitro-Untersuchungen bewertet; dies gilt nicht für Stoffe zur Beherrschung einer Kontamination mit Radionukliden. Die für die einzelnen Funktionsgruppen geeigneten Endpunkte sind in der nachfolgenden Tabelle aufgeführt.
Endpunkte für verschiedene technologische Zusatzstoffe
| Funktionsgruppe | Endpunkte für den Nachweis der Wirksamkeit | |
| a) | Konservierungsmittel | Hemmung des mikrobiellen Wachstums, insbesondere des Wachstums von biotischen Organismen und Verderbnisorganismen. Der Zeitraum, während dessen laut Antrag eine konservierende Wirkung vorliegt, wird nachgewiesen. |
| b) | Antioxidationsmittel | Schutz gegen oxidative Schädigungen der wichtigsten Nährstoffe bzw. Bestandteile während der Verarbeitung und/oder Lagerung von Futtermitteln. Der Zeitraum, während dessen laut Antrag eine schützende Wirkung vorliegt, wird nachgewiesen. |
| c) | Emulgatoren | Bildung bzw. Aufrechterhaltung stabiler Emulsionen, die aus eigentlich nicht mischbaren oder kaum mischbaren Futtermittelbestandteilen bestehen |
| d) | Stabilisatoren | Aufrechterhaltung der physikalischchemischen Beschaffenheit von Futtermitteln |
| e) | Verdickungsmittel | Viskosität der Futtermittelausgangserzeugnisse oder Futtermittel |
| f) | Geliermittel | Bildung eines Gels, wodurch sich die Textur des Futtermittels ändert |
| g) | Bindemittel | Härte der Pellets, Verlauf der Pelletierung |
| h) | Stoffe zur Beherrschung einer Kontamination mit Radionukliden | Nachweis der verringerten Kontamination von Lebensmitteln tierischen Ursprungs |
| i) | Trennmittel | Fließfähigkeit. Der Zeitraum, während dessen laut Antrag eine trennende Wirkung vorliegt, wird nachgewiesen. |
| j) | Säureregulatoren | pH-Wert und/oder Pufferkapazität in Futtermitteln |
| k) | Silierzusatzstoffe | - Verbesserte Silageerzeugung;
- Hemmung unerwünschter Mikroorganismen; - Verringerung des Sickersaftes; - erhöhte aerobe Stabilität |
Silierzusatzstoffe
Zum Nachweis der im Antrag erwähnten Wirkung auf den Vorgang des Silierens 2 werden gesonderte Untersuchungen durchgeführt. Die Untersuchungen werden an einer Probe jeder der folgenden Kategorien vorgenommen (wenn es um alle oder unbestimmte Arten von Futtermitteln geht):
Beschränkt sich ein Antrag auf Unterkategorien von Futtermitteln, die anhand der Trockenmasse beschrieben werden, wird der Trockenmassebereich ausdrücklich angeführt. Es werden drei Untersuchungen mit Material durchgeführt, die diesen Bereich repräsentieren; falls möglich, werden dazu Proben unterschiedlichen pflanzlichen Ursprungs verwendet.
Für die einzelnen Futtermittel sind eigene Untersuchungen erforderlich.
Die Untersuchungen dauern in der Regel 90 Tage oder länger und werden bei einer gleichmäßigen Temperatur durchgeführt (empfohlen wird der Bereich zwischen 15 °C und 25 °C). Ein kürzerer Zeitraum muss begründet werden.
Im Allgemeinen werden die folgenden Parameter im Vergleich zur negativen Kontrolle gemessen:
Zusätzlich werden zur Untermauerung der vorgesehenen Wirkung gegebenenfalls andere mikrobiologische und chemische Parameter bewertet (z.B. Anzahl der Hefearten, die Lactate assimilieren, Anzahl der Clostridien, Listerien sowie biogene Amine).
Eine gewünschte Wirkung zur Verringerung des Sickersaftes wird anhand der gesamten Abflussmenge bewertet, die während des ganzen Untersuchungszeitraums entsteht; hierbei wird die zu erwartende Auswirkung auf die Umwelt (z.B. Ökotoxizität des Sickersaftes oder biochemischer Sauerstoffbedarf) berücksichtigt. Die Verringerung des Sickersaftes wird direkt nachgewiesen. Das Silo muss eine derartige Kapazität haben, dass sich Sickersaft unter Anwendung von Druck freisetzen lässt. Die Untersuchung dauert in der Regel 50 Tage. Ein davon abweichender Zeitraum wird begründet.
Eine erhöhte aerobe Stabilität wird im Vergleich mit einer negativen Kontrolle belegt. Untersuchungen zur Stabilität werden nach der Exposition gegenüber Luft für mindestens sieben Tage durchgeführt; der Zusatzstoff soll im Vergleich zu einer negativen Kontrolle nachweislich zu einer mindestens zwei Tage längeren Stabilität. Führen. Es wird empfohlen, den Versuch bei einer Umgebungstemperatur von 20 °C und einem Temperaturanstieg von 3 °C oder mehr gegenüber der Umgebungstemperatur durchzuführen; der Temperaturanstieg wird als Anzeichen von Instabilität betrachtet. Anstelle der Temperatur kann die CO2-Produktion gemessen werden.
1.5 Abschnitt V: Plan zur marktbegleitenden Beobachtung
Dieser Abschnitt kommt gemäß Artikel 7 Absatz 3 Buchstabe g der Verordnung (EG) Nr. 1831/2003 zur Anwendung. Das heißt, ein Plan zur marktbegleitenden Beobachtung ist nur bei Zusatzstoffen erforderlich, die aus GVO bestehen oder daraus hergestellt wurden.
|
weiter . | |
(Stand: 30.11.2020)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: 90.- € netto (Grundlizenz)
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion