

umwelt-online: Entscheidung 2002/657/EG Durchführung von Analysemethoden und die Auswertung von Ergebnissen (3)
| |
zurück | |
3.1.1.5. Kalibrierkurven
Wenn Kalibrierkurven zur Quantifizierung verwendet werden, gilt Folgendes:
Wenn eine serielle Kalibrierung auf der Basis einer Standardlösung erforderlich ist, müssen zulässige Bereiche für die Parameter der Kalibrierkurve, die von Serie zu Serie schwanken können, angegeben werden.
3.1.2. Herkömmliche Validierungsverfahren
Für die Berechnung der Parameter gemäß den herkömmlichen Methoden müssen mehrere Einzeluntersuchungen durchgeführt werden. Jedes Leistungsmerkmal muss für jede größere Änderung bestimmt werden (siehe unter Anwendbarkeit/Robustheit weiter oben). Bei Multianalytmethoden können mehrere Analyte gleichzeitig analysiert werden, sofern mögliche relevante Störungen zuvor ausgeschlossen werden. Mehrere Leistungsmerkmale können auf ähnliche Weise bestimmt werden. So empfiehlt es sich, zur Reduzierung des Aufwands die Untersuchungen soweit wie möglich zu kombinieren (z.B. Wiederholpräzision und laborinterne Reproduzierbarkeit mit der Spezifität, die Analyse von Leerwertproben zur Bestimmung der Entscheidungsgrenze und die Prüfung auf Spezifität).
3.1.2.1. Wiederfindung
Wenn kein zertifiziertes Referenzmaterial (CRM) zur Verfügung steht, muss die Wiederfindung durch Untersuchungen mit dotierter Leerwertmatrix bestimmt werden, z.B. nach folgendem Plan:
Diese herkömmliche Methode der Bestimmung der Wiederfindung ist eine Variante der im Abschnitt 3.5 beschriebenen Standardadditionsmethode, wenn
Wenn eine der oben genannten Bedingungen nicht (oder vermutlich nicht) herzustellen ist, muss das im Abschnitt 3.5 beschriebene vollständige Verfahren zur Bestimmung der Wiederfindung mit der Methode der Standardaddition durchgeführt werden.
3.1.2.2. Wiederholpräzision
3.1.2.3. Laborinterne Reproduzierbarkeit
3.1.2.4. Reproduzierbarkeit
Wenn die Reproduzierbarkeit verifiziert werden muss, sollten die Laboratorien an Methodenvergleichsstudien gemäß ISO 5725-2 teilnehmen (5).
3.1.2.5. Entscheidungsgrenze (CCα)
Die Entscheidungsgrenze muss gemäß den Anforderungen für die Identifizierung oder die Identifizierung plus Quantifizierung, die unter "Leistungskriterien und sonstige Anforderungen für Analysemethoden" (Teil 2) definiert sind, festgelegt werden.
Im Fall von Stoffen, für die kein zulässiger Grenzwert festgelegt worden ist, kann die Entscheidungsgrenze CCα wie folgt bestimmt werden:
Im Fall von Stoffen mit einem festgelegten zulässigen Grenzwert kann die Entscheidungsgrenze CCα wie folgt bestimmt werden:
Siehe auch Artikel 5 und Abschnitt 3.2.
3.1.2.6. Nachweisvermögen (CCβ)
Das Nachweisvermögen sollte gemäß den festgelegten Anforderungen für das Screening, die Identifizierung oder die Identifizierung plus Quantifizierung (siehe Teil 2) bestimmt werden.
Im Fall von Stoffen, für die kein zulässiger Grenzwert festgelegt worden ist, kann das Nachweisvermögen CCß wie folgt bestimmt werden:
Im Fall von Stoffen, für die ein zulässiger Grenzwert festgelegt worden ist, kann das Nachweisvermögen CCβ wie folgt bestimmt werden:
Siehe auch Abschnitt 3.2.
3.1.2.7. Robustheit (größere Änderungen)
Die Analysenmethode sollte unter unterschiedlichen Versuchsbedingungen, unter anderem zum Beispiel verschiedene Spezies, verschiedene Matrices oder verschiedene Probennahmebedingungen, geprüft werden. Die vorgenommenen Änderungen sollten wesentlich sein. Die Bedeutung dieser Änderungen kann beispielsweise mit der Methode von Youden beurteilt werden (15) (16). Jedes Leistungsmerkmal sollte für alle größeren Änderungen bestimmt werden, die nachweislich einen wesentlichen Einfluss auf die Leistungsfähigkeit des Tests haben.
3.1.3. Validierung nach alternativen Modellen
Wenn alternative Validierungsverfahren praktiziert werden, müssen das zugrunde liegende Modell und die Validierungsstrategie sowie die jeweiligen Voraussetzungen, Annahmen und Formeln im Validierungsplan dargelegt oder zumindest muss auf die entsprechenden Quellen verwiesen werden. Im Folgenden wird ein Beispiel für einen alternativen Validierungsansatz gegeben. Wenn z.B. das Modell der laborinternen Validierung angewandt wird, werden die Leistungsmerkmale so bestimmt, dass im Rahmen desselben Validierungsverfahrens eine Validierung für größere Änderungen möglich ist. Hierzu muss ein Versuchsplan für die Validierung erstellt werden.
3.1.3.1. Versuchsplan
Ein Versuchsplan muss erstellt werden, der die Anzahl der verschiedenen Tierarten und der verschiedenen untersuchten Faktoren berücksichtigt. Deshalb muss im ersten Schritt des Validierungsverfahrens geprüft werden, welche Probenpopulationen künftig im Laboratorium analysiert werden, um die wichtigsten Tierarten und jene Faktoren auswählen zu können, welche die Messergebnisse beeinflussen können. Anschließend muss der Konzentrationsbereich zweckbezogen entsprechend dem interessierenden Wert gewählt werden.
Beispiel:
Tabelle 13Beispiele für Faktoren, die für ein Validierungsverfahren als wichtig erachtet werden
| Geschlecht des Tieres | (Faktor 1) |
| Rasse | (Faktor 2) |
| Transportbedingungen | (Faktor 3) |
| Lagerungsbedingungen | (Faktor 4) |
| Frische der Probe | (Faktor 5) |
| Mastbedingungen | (Faktor 6) |
| Verschiedene Analytiker mit unterschiedlicher Erfahrung | (Faktor 7) |
Tabelle 14Möglicher Versuchsplan für das oben genannte Beispiel
| Tierart | Faktor 1 | Faktor 2 | Faktor 3 | Faktor 4 | Faktor 5 | Faktor 6 | Faktor 7 | Probe Nr. |
| A | + | + | + | + | - | + | - | 1 |
| A | + | + | - | - | + | - | - | 2 |
| A | + | - | + | - | - | - | + | 3 |
| A | + | - | - | + | + | + | + | 4 |
| A | - | + | + | - | + | + | + | 5 |
| A | - | + | - | + | - | - | + | 6 |
| A | - | - | + | + | + | - | - | 7 |
| A | - | - | - | - | - | + | - | 8 |
| B | + | + | + | + | + | - | + | 9 |
| B | + | + | - | - | - | + | + | 10 |
| B | + | - | + | - | + | + | - | 11 |
| B | + | - | - | + | - | - | - | 12 |
| B | - | + | + | - | - | - | - | 13 |
| B | - | + | - | + | + | + | - | 14 |
| B | - | - | + | + | - | + | + | 15 |
| B | - | - | - | - | + | - | + | 16 |
Da jede Probe (jede Kombination von Faktorausprägungen) mit 4 verschiedenen Konzentrationen im Bereich des interessierenden Werts dotiert und eine Leerwertprobe für jede Kombination analysiert werden muss, sind 5 x 16 = 80 Analysen für die gesamte Validierungsstudie durchzuführen.
Aus diesen 80 Messergebnissen können folgende Leistungsmerkmale berechnet werden (13) (14).
Wiederfindung
Diese Leistungsmerkmale erlauben die umfassende Bewertung des Leistungsgrads der Methode, da nicht nur der Einfluss der einzelnen Faktoren untersucht wird, sondern auch derjenige der einschlägigen Kombinationen dieser Faktoren. Mit Hilfe dieses Versuchsdesigns kann festgestellt werden, ob irgendeiner der gewählten Faktoren aus der Gesamtkalibrierkurve ausgeschlossen werden muss, weil er wesentlich von den Standardabweichungen der anderen Faktoren abweicht.
3.1.3.2. Gütekurve
Die Gütekurve gibt Aufschluss über das Nachweisvermögen der Methode innerhalb des gewählten Konzentrationsbereichs. Sie bezieht sich auf die β -Fehlerwahrscheinlichkeit beim Einsatz der untersuchten Methode. Die Gütekurve erlaubt die Berechnung des Nachweisvermögens für die jeweiligen Kategorien (Screening, Bestätigung) oder Arten (qualitativ oder quantitativ) von Methoden für einen bestimmten β-Fehler (z.B. 5 %).
Abbildung 1 Gütekurve

Abbildung 1 zeigt ein Beispiel für die grafische Darstellung des Nachweisvermögens (CCβ) einer Analysemethode. Für die betreffende Methode besteht bei einer Konzentration von 0,50 µg/kg eine Restwahrscheinlichkeit von 5 %, dass eine falsche Entscheidung getroffen wird. Bei einer Konzentration von 0,55 µ/kg nimmt die Wahrscheinlichkeit einer falsch negativen Entscheidung auf 1 % ab.
3.1.3.3. Reproduzierbarkeit
Die Bestimmung der Reproduzierbarkeit einer Methode nach dem Konzept der laborinternen Validierung erfordert die wiederholte Teilnahme an Laboreignungsprüfungen gemäß ISO-Leitfaden 43-1 (3) und 43-2 (4). Die Laboratorien können ihre Methoden selbst wählen, sofern diese Methoden unter Routinebedingungen eingesetzt werden. Die Standardabweichung des Laboratoriums kann zur Bewertung der Reproduzierbarkeit der Methode herangezogen werden.
3.2. Grafische Darstellung der verschiedenen analytischen Grenzen
Abbildung 2 Stoffe, für die kein zulässiger Grenzwert festgelegt worden ist
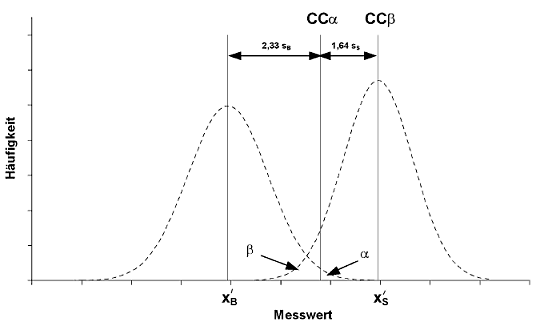
| XxS | mittlerer Messwert der verunreinigten Probe |
| SB | Standardabweichung der Leerwertprobe (bestimmt unter laborinternen Reproduzierbarkeitsbedingungen) |
| SS | Standardabweichung der verunreinigten Probe (bestimmt unter laborinternen Reproduzierbarkeitsbedingungen) |
| α | Rate falsch positiver Ergebnisse |
| β | Rate falsch negativer Ergebnisse |
| CCα | Messwert reit einem gegebenen α-Fehler und 50 %-β-Fehler |
| CCβ | Messwert mit einem sehr kleinen α-Fehler und einem gegebenen β-Fehler |
Abbildung 3 Stoffe mit einem festgelegten zulässigen Grenzwert
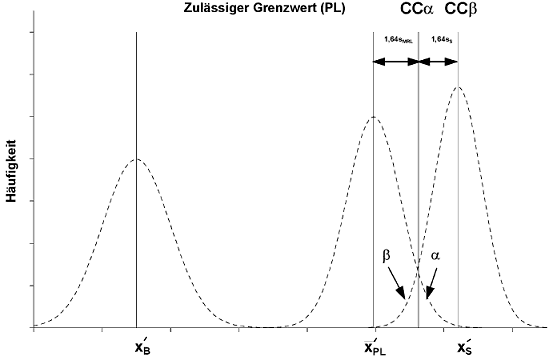
| XxB | mittlere "Konzentration" der Leerwertprobe |
| XxZUL. GRENZWERT | mittlere Konzentration der Probe, die den Analyten in der Konzentration des zulässigen Grenzwerts enthält |
| XxS | mittlere Konzentration der verunreinigten Probe |
| SZUL. GRENZWERT | Standardabweichung der Probe, die den Analysen in der Konzentration des zulässigen Grenzwerts enthält (bestimmt unter laborinternen Reproduzierbarkeitsbedingungen) |
| SS | Standardabweichung der verunreinigten Probe (bestimmt unter laborinternen Reproduzierbarkeitsbedingungen) |
| α | Rate falsch positiver Ergebnisse |
| β | Rate falsch negativer Ergebnisse |
| CCα | Messwert mit einem gegebenen α-Fehler und 50 %-β-Fehler |
| CCβ | Messwert mit einem sehr kleinen (α-Fehler und einem gegebenen β-Fehler |
3.3. Berechnungsbeispiel für einen Robustheitstest gegenüber geringfügigen Änderungen nach dem Ansatz von Youden (16)
Vergleich der Mittelwerte (A)
| AA = | Σ (Ai)/4 | Die Mittelwerte der Großbuchstaben (Aa bis AG mit den Mittelwerten ihrer entsprechenden Kleinbuchstaben (Aa bis Ag) vergleichen. Wenn ein Faktor eine Auswirkung hat, ist der Unterschied wesentlich größer als die Unterschiede der anderen Faktoren.
Eine robuste Methode sollte nicht durch Veränderungen beeinflusst werden, die fast sicher zwischen Laboratorien zu beobachten sind. Wenn kein auffallender Unterschied besteht, ist das reellste Maß des Zufallsfehlers durch die sieben Unterschiede angegeben. |
| AB = | Σ (Bi)/4 | |
| AC = | Σ (Ci)/4 | |
| AD = | Σ (Di)/4 | |
| AE = | Σ (Ei)/4 | |
| AF = | Σ (Ei)/4 | |
| AG = | Σ (Gi)/4 | |
| Aa = | Σ (ai)/4 | |
| Ab = | Σ (bi)/4 | |
| Ac = | Σ (ci)/4 | |
| Ad = | Σ (di)/4 | |
| Ae = | Σ (ei)/4 | |
| Af = | Σ (fi)/4 | |
| Ag= | Σ (gi)/4 |
| Unterschiede (Di) | Unterschiedsquadrate (Di2) |
| Da = a - a = Σ(Ai) - Σ(ai) | Da2 = Wert a |
| Db = B - b = Σ(Bi) - Σ(bi) | Db2 = Wert b |
| Dc = C - c = Σ(Ci) - Σ(ci) | Dc2 = Wert c |
| Dd = D - d = Σ(Di) - Σ(di) | Dd2 = Wert d |
| De = E - e = Σ(Fi) - Σ(ei) | De2 = Wert e |
| Df = F - f = Σ(Fi) - Σ(fi) | Df2 = Wert f |
| Dg = G - g = Σ(Gi) - Σ(gi) | Dg2 = Wert g |
Standardabweichung der Unterschiede Di (SDi)
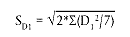
Wenn SD wesentlich größer als die Standardabweichung der unter laborinternen Reproduzierbarkeitsbedingungen durchgeführten Methode (siehe oben) ist, so steht von vornherein fest, dass alle Faktoren zusammen eine Auswirkung auf das Ergebnis haben, auch wenn jeder einzelne Faktor keinen wesentlichen Einfluss zeigt, und dass die Methode somit nicht robust genug gegenüber den gewählten Veränderungen ist.
3.4. Berechnungsbeispiele für das laborinterne Validierungsverfahren
Beispiele und Berechnungen für das laborinterne Validierungsmodell, das im Abschnitt 3.1.3, Validierung nach alternativen Modellen, beschrieben ist (13) (14).
3.5. Beispiele für die Standardadditionsmethode
Eine Untersuchungsprobe mit einem Gehalt T des Analyten wird in zwei Analysenproben 1 und 2 mit den Massen m, und m2 geteilt. Die Analysenprobe 2 wird mit einem Volumen VA einer Lösung der Konzentration pa des Analyten dotiert. Zwei Extrakte der Analysenproben mit den Volumina V1 und V2 werden nach den Extraktions- und Aufreinigungsschritten der Methode gewonnen. Die Wiederfindung des Analyten soll rc sein. Beide Extrakte werden mit einer Messmethode mit der Empfindlichkeit b gemessen und liefern ein Analysenergebnis von x1 bzw. x2.
Angenommen rc und b sind gleich für den Analyten in der nativen Probe und in der dotierten Probe, dann lässt sich der Gehalt T wie folgt berechnen:
| X1⋅V1⋅ ρ A⋅VA | |
| T = | |
| (x2⋅V2⋅m1- x1⋅V1⋅m2) |
Die Methode erlaubt die Bestimmung der Wiederfindung rc. Dabei wird, zusätzlich zum oben beschriebenen Test, ein Teil des Extrakts der Analysenprobe 1 (Volumen V3) mit einer bekannten Menge ρB⋅VB des Analyten dotiert und analysiert. Das Analysenergebnis ist x3 und die Wiederfindung ist:
| x2⋅V1⋅V2⋅ ρ B⋅VB | |
| rc = | |
| [x3⋅V1⋅V3(T.m2 + ρA⋅VA) - x2- V2⋅T.m1(V3-VB)] |
Außerdem kann die Empfindlichkeit b wie folgt berechnet werden:
| x1⋅V1 | |
| b = | |
| rc.T.m1 |
Alle Anwendungsbedingungen und die Einzelheiten sind bereits beschrieben worden (18).
4. Verwendete Abkürzungen
| AAS | Atomabsorptionsspektrometrie |
| AES | Atomemissionsspektrometrie |
| AOAC-I | Association of Official Analytical Chemists INTERNATIONAL |
| B | gebundene Fraktion (Immunoassays) |
| CI | chemische Ionisierung |
| CRM | zertifiziertes Referenzmaterial |
| CV | Variationskoeffizient |
| 2D | zweidimensional |
| DAD | Dioden-Array-Detektion |
| DPASV | differenzielle anodische Stripping-Pulsvoltammetrie |
| ECD | Elektroneneinfangdetektion |
| EI | Elektronenstoßionisierung |
| GC | Gaschromatografie |
| HPLC | Hochleistungsflüssigchromatografie |
| HPTLC | Hochleistungsdünnschichtchromatografie |
| HRMS | hochauflösende Massenspektrometrie |
| ICP-AES | Atomemissionsspektrometrie mit induktiv gekoppeltem Plasma |
| ICP-MS | Massenspektrometrie mit induktiv gekoppeltem Plasma |
| IR | infrarot |
| ISO | International Standard Organisation |
| LC | Flüssigchromatografie |
| LRMS | niedrig auflösende Massenspektrometrie |
| MRPL | geforderte Mindestleistungsgrenze |
| MS | Massenspektrometrie |
| m/z | Masse-Ladungs-Verhältnis |
| RF | Retentionsfaktor (TLC) |
| RSDL | relative Standardabweichungen des Laboratoriums |
| SIM | Einzelmassenregistrierung |
| TLC | Dünnschichtchromatografie |
| UV | ultraviolettes Licht |
| VIS | sichtbares Licht |
5. Literatur
(1) ISO 17025: 1999 General requirement for the competence of calibration and testing laboratories
(2) ISO 3534-1: 1993 Statistics - Vocabulary and symbols - Part 1: Probability and general statistical terms
(3) ISO Guide 43-1:1997 Proficiency testing by interlaboratory comparisons - Part 1: Development and operation of proficiency testing schemes
(4) ISO Guide 43-2:1997 Proficiency testing by interlaboratory comparisons - Part 2: Selection and use of proficiency testing schemes by laboratory accreditation bodies
(5) ISO 5725:1994 Accuracy (trueness and precision) of measurement methods and results - Part 1: General principles and definitions; ISO 5725-2 Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method; Part 4: Basic methods for the determination of the trueness of a standard measurement method
(6) ISO 78-2:1999 Chemistry - Layouts for Standards - Part 2: Methods of chemical analysis
(7) W. G de Ruig and J. M Weseman "a new approach to confirmation by infrared spectrometry"). Chemometrics 4 (1990) 61-77.
(8) See e.g. May, T. W., Brumbaugh, W.G., 1982, Matrix modifier and L'vov platform for ehmination of matrix interferences in the analysis of fish tissues for lead by graphite furnace atomic absorption spectrometry: Analytical Chemistry 54(7):1032-1037 (90353)
(9) Applications of Zeeman Graphite Furnace Atomic Absorption Spectrometry in the Chemical Laboratory and in Toxicology, C. Minoia, S. Caroli (Eds.), Pergamon Press (Oxford), 1992, pp. xxvi + 675
(10) Inductively Coupled Plasmas in Analytical Atomic Spectrometry, A. Montaser, D. W. Golighty (Eds.), VCH Publishers, Inc. (New York), 1992
(11) Plasma Source Mass Spectrometry Developments and Applications, G. Holland, S. D. Tanner (Eds.), The Royal Society of Chemistry, 1997, pp. 329
(12) IUPAC (1995), Protocol for the design, conduct and interpretation of method-performance studies, Pure & Applied Chem, 67, 331
(13) Jülicher, B., Gowik, P. und Uhlig, S. (1998) Assessment of detection methods in trace analysis by means of a statisticahy based in-house validation concept. Analyst, 120, 173
(14) Gowik, P., Jülicher, B. und Uhlig, S. (1998) Multi-residue method for non-steroidal anti-inflammatory drugs in plasma using high performance liquid chromatography-photodiode-array detection. Method description and comprehensive in-house validation.). Chromatogr., 716, 221
(15) OAC-I Peer Verified Methods, Policies and Procedures, 1993, ADAC International, 2200 Wilson Blvd., Suite 400, Arlington, Virginia 22201-3301, USA
(16) W. J. Youden; Steiner, E. H.; "Statistical Manual of the ADAC-Association of Official Analytical Chemists",ADAC-I, Washington DC: 1975, p. 35 ff
(17) ISO 11843: 1997 Capabflity of detection - Part 1: Terms and definitions, Part 2: Methodology in the linear calibration case
(18) R. W. Stephany & L. A. van Ginkel: "Yield or recovery: a world of difference". Proceedings 8th Euro Food Chem, Vienna, Austria September 18-20 (1995) Federation of European Chemical Societies, Event 206. ISBN 3-90055417X, page 2-9
(19) Council Directive 71/354/EEC of 18 October 1971 an the approximation of the laws of the Member States relating to units of measurement Official Journal L 243, 29.10.1971, p. 29
(20) ISO 31-0:1992 Quantities and units - Part 0: General principles
________________________________
1) Ausbeute: der Massenanteil des in der Probe enthaltenen Analyten, der im Endextrakt vorhanden ist.
2) Wiederfindung (hier): der Massenanteil des der Probe zugesetzten Analyten, der im Endextrakt vorhanden ist. Im weiteren Verlauf des Dokuments wird vorausgesetzt, dass Ausbeute und Wiederfindung gleich sind, weshalb nur der Begriff "Wiederfindung" verwendet wird.
| Mindestleistungsgrenzen | Anhang II |
| Stoff und/oder Metabolit | Matrix | MRPL |
| Chloramphenicol | Fleisch Eier Milch Urin Erzeugnisse der Aquakultur Honig |
0,3 µg/kg |
| Medroxy-Progesteron-Acetat | Schweinenierenfett | 1 µg/kg |
Nitrofuranmetaboliten:
|
Geflügelfleisch Erzeugnisse der Aquakultur |
1 µg/kg für alle |
| Summe von Malachit- und Leukomalachitgrün | Fleisch von Erzeugnissen der Aquakultur | 2 µg/kg |
| |
ENDE | |
(Stand: 26.05.2021)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: ab 105.- € netto
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion