

umwelt-online: Verordnung (EG) Nr. 152/2009 zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln (2)
|
zurück | |
5. Verfahren
5.1 Vorbereitung der Probe
Die Probe wird so fein vermahlen, dass sie ein Sieb mit 0,5 mm Maschenweite passieren kann. Proben mit hohem Feuchtigkeitsgehalt müssen vor dem Vermahlen entweder bei einer Temperatur von höchstens 50 °C luftgetrocknet oder aber gefriergetrocknet werden. Proben mit hohem Fettgehalt sind vor dem Vermahlen mit Petrolether ( 3.12) zu extrahieren.
5.2 Bestimmung des Gehalts an freien Aminosäuren in Futtermitteln und Vormischungen
Eine geeignete Menge (1 bis 5 g) der vorbereiteten Probe ( 5.1) auf 0,2 mg genau in einen Erlenmeyerkolben einwiegen und 100,0 ml Extraktionslösung ( 3.21) hinzufügen. Mit einem mechanischen Schüttler 60 min schütteln bzw. mit einem Magnetrührer (4.8) rühren. Nach dem Absetzen lassen werden mit einer Pipette 10,0 ml der überstehenden Lösung in ein 100-ml-Becherglas gegeben.
Es werden 5,0 ml Sulfosalicylsäurelösung ( 3.22) unter Rühren hinzugefügt, und mithilfe des Magnetrührers wird 5 min weitergerührt. Die überstehende Lösung wird filtriert oder zentrifugiert, um alle Ausfällungen zu entfernen. Von der klaren Lösung werden 10,0 ml in ein 100-ml-Becherglas gegeben, und unter Verwendung von Natriumhydroxidlösung ( 3.18) wird der pH-Wert auf 2,20 eingestellt. Die Lösung wird mit Citratpufferlösung ( 3.24) in einen Messkolben geeigneter Größe überführt und mit Citratpufferlösung ( 3.24) zur Marke aufgefüllt.
Bei Verwendung eines internen Standards werden vor dem Auffüllen 1,00 ml der Stammlösung des internen Standards ( 3.27.3) für jeweils 100 ml Endlösung hinzugefügt und mit Citratpufferlösung ( 3.24) zur Marke aufgefüllt.
Anschließend wird die Chromatografie gemäß 5.4 durchgeführt.
Werden die Extrakte nicht am selben Tag chromatografiert, sind sie bei < 5 °C aufzubewahren.
5.3 Bestimmung des Gehalts an Gesamtaminosäuren
5.3.1 Oxidation
0,1 bis 1 g der vorbereiteten Probe ( 5.1) werden auf 0,2 mg genau eingewogen
Die Einwaage muss einen Stickstoffgehalt von etwa 10 mg und einen Feuchtigkeitsgehalt von höchstens 100 mg haben.
Der Kolben/die Flasche wird in ein Eisbad gestellt und auf 0 °C abgekühlt. Dann werden 5 ml der Oxidationsmischung ( 3.23) hinzugefügt, und es wird mithilfe eines Glasspatels mit gebogener Spitze gemischt. Der Behälter, der den Spatel enthält, wird mit einer luftdichten Folie verschlossen, das Eiswasserbad mit dem verschlossenen Behälter in einen Kühlschrank mit einer Temperatur von 0 °C gestellt und 16 h stehen gelassen. Nach 16 h wird der Behälter aus dem Kühlschrank genommen und das überschüssige Oxidationsreagenz durch Zusatz von 0,84 g Natriumdisulfit ( 3.4) zersetzt.
Es wird gemäß 5.3.2.1 weiterverfahren.
5.3.2 Hydrolyse
5.3.2.1 Hydrolyse der oxidierten Proben
Zu der oxidierten Probe (die gemäß 5.3.1 vorbereitet wurde) werden 25 ml Hydrolysemischung ( 3.20) gegeben. Dabei ist darauf zu achten, dass alle Probenrückstände, die an den Seitenwänden des Gefäßes sowie am Spatel haften, abgewaschen werden.
Je nach dem verwendeten Hydrolyseverfahren wird gemäß 5.3.2.3 oder 5.3.2.4 weiterverfahren.
5.3.2.2 Hydrolyse der nicht oxidierten Proben
In einen 100-ml- oder 250-ml-Rundkolben ( 4.1) bzw. in eine 100-ml-Flasche mit Schraubverschluss ( 4.2) werden 0,1 bis 1 g der vorbereiteten Probe ( 5.1) auf 0,2 mg genau eingewogen. Die Einwaage muss einen Stickstoffgehalt von ca. 10 mg aufweisen. Vorsichtig 25 ml Hydrolysemischung ( 3.20) hinzufügen und mit der Probe vermischen. Es wird entweder gemäß 5.3.2.3 oder gemäß 5.3.2.4 weiterverfahren.
5.3.2.3 Offene Hydrolyse (Rückflusshydrolyse)
Zu der nach 5.3.2.1 oder 5.3.2.2 hergestellten Mischung werden 3 Glasperlen hinzugegeben. Sie wird zum Sieden erhitzt und unter Rückfluss genau 23 h am Sieden gehalten. Nach Beendigung der Hydrolyse wird der Rückflusskühler mit 5 ml Citratpufferlösung ( 3.24) gespült und der abgehängte Kolben im Eisbad gekühlt.
Es wird gemäß 5.3.3 weiterverfahren.
5.3.2.4 Geschlossene Hydrolyse
Die Flasche mit der nach 5.3.2.1 oder 5.3.2.2 vorbereiteten Mischung wird bei 110 °C in den Trockenschrank ( 4.3) gestellt. Um einen durch Gasentwicklung hervorgerufenen Druckaufbau zu vermeiden und eine Explosion zu verhindern, wird der Schraubverschluss während der ersten Stunde nur lose auf die Flasche gelegt. Die Flasche darf nicht verschlossen werden. Nach 1 h wird die Flasche mit dem Deckel fest verschlossen und 23 h im Trockenschrank ( 4.3) belassen. Nach Beendigung der Hydrolyse wird die Flasche aus dem Trockenschrank genommen, der Verschluss vorsichtig geöffnet, die Flasche in ein Eisbad gestellt und abkühlen gelassen.
Je nach dem Verfahren für die pH-Wert-Einstellung ( 5.3.3) wird der Inhalt der Flasche quantitativ mit Citratpufferlösung ( 3.24) in ein 250-ml-Becherglas oder in einen 250-ml-Rundkolben überführt.
Es wird gemäß 5.3.3 weiterverfahren.
5.3.3 pH -Wert - Einstellung
Je nach der Natriumionentoleranz des Aminosäureanalysators ( 4.9) wird die pH-Wert-Einstellung gemäß 5.3.3.1 oder 5.3.3.2 vorgenommen.
5.3.3.1 Für Aminosäure analysatoren oder HPLC - Einrichtungen ( 4.9), die eine niedrige Natriumionenkonzentration erfordern
Werden Aminosäureanalysatoren verwendet, bei denen eine geringe Natriumionenkonzentration erforderlich ist (wenn also das Säurevolumen reduziert werden muss), wird die Verwendung einer Stammlösung eines internen Standards ( 3.27.3) empfohlen.
In diesem Fall sind dem Hydrolysat vor dem Verdampfen 2,00 ml der internen Standardlösung ( 3.27.3) hinzuzufügen.
Zu dem nach 5.3.2.3 oder 5.3.2.4 erhaltenen Hydrolysat werden 2 Tropfen 1-Octanol ( 3.15) gegeben.
Das Volumen wird am Rotationsverdampfer ( 4.7) unter Vakuum bei 40 °C auf 5 bis 10 ml reduziert. Wurde das Volumen beim Einengen versehentlich auf weniger als 5 ml reduziert, wird das Hydrolysat verworfen und die Analyse wiederholt.
Der pH-Wert wird mit Natriumhydroxidlösung ( 3.18) auf 2,20 eingestellt, und es wird gemäß 5.3.4 weiterverfahren.
5.3.3.2 Für alle sonstigen Aminosäureanalysatoren ( 4.9)
Die nach 5.3.2.3 oder 5.3.2.4 erhaltenen Hydrolysate werden durch vorsichtige Zugabe unter Rühren von 17 ml Natriumhydroxidlösung ( 3.17) teilweise neutralisiert, wobei eine Temperatur von unter 40 °C eingehalten werden muss.
Der pH-Wert wird mit Natriumhydroxidlösung ( 3.17 und anschließend 3.18) bei Raumtemperatur auf 2,20 eingestellt, und es wird gemäß 5.3.4 weiterverfahren.
5.3.4 Probenlösung für die Chromatografie
Das auf pH 2,20 eingestellte Hydrolysat ( 5.3.3.1 oder 5.3.3.2) wird mit Citratpufferlösung ( 3.24) quantitativ in einen 200-ml-Messkolben überführt und mit Citratpufferlösung ( 3.24) zur Marke aufgefüllt.
Falls bisher noch kein interner Standard hinzugefügt worden ist, werden vor dem Auffüllen 2,00 ml des internen Standards ( 3.27.3) hinzugegeben und dann mit Citratpufferlösung ( 3.24) zur Marke aufgefüllt. Es wird sorgfältig gemischt.
Anschließend wird die Chromatografie gemäß 5.4 durchgeführt.
Falls die Probenlösungen nicht am selben Tag chromatografiert werden, sind sie bei < 5 °C aufzubewahren.
5.4 Chromatografie
Falls erforderlich, wird der Extrakt ( 5.2) oder das Hydrolysat (5.3.4) vor der Chromatografie auf Raumtemperatur gebracht. Die Mischung wird geschüttelt und eine geeignete Menge durch einen 0,22-m-Membranfilter ( 4.5) filtriert. Die so erhaltene klare Lösung wird der Ionenaustauschchromatografie unter Verwendung eines Aminosäureanalysators bzw. einer HPLC-Einrichtung ( 4.9) unterzogen.
Die Einspritzung kann von Hand oder automatisch erfolgen. Wesentlich ist, dass jeweils die gleiche Menge ± 0,5 % der Standardlösung und der Probenlösung eingespritzt wird, es sei denn, es wird ein interner Standard verwendet. Das Verhältnis der Konzentrationen von Natriumionen und Aminosäuren sollte in der Standard- und der Probenlösung so ähnlich wie möglich sein.
Die Häufigkeit von Kalibrationsläufen hängt von der Stabilität des Ninhydrinreagenzes und des Analysatorsystems ab. Die Standardlösung oder die Probenlösung wird mit Citratpufferlösung ( 3.24) so verdünnt, dass die Peakfläche des Standards etwa 30 bis 200 % der Peakfläche der einzelnen Aminosäuren in der Probenlösung ausmacht.
Die Chromatografie der Aminosäuren unterscheidet sich leicht je nach Art des verwendeten Analysators und des eingesetzten Harzes. Das System muss in der Lage sein, die Aminosäuren vollständig voneinander und von den sonstigen Ninhydrinpositiven Substanzen zu trennen. Im Betriebsbereich muss das chromatografische System bei Änderungen der Aminosäuremengen, die der Säule hinzugefügt wird, eine lineare Reaktion aufweisen.
Wird eine equimolare Lösung der zu bestimmenden Aminosäuren analysiert, sollten bei der Chromatografie die unten angegebenen Verhältnisse von Tal zu Peakhöhe zwischen den überlappenden Peaks erreicht werden. Diese equimolare Lösung muss mindestens 30 % der maximalen Menge jeder Aminosäure enthalten, die mit dem jeweiligen Aminosäureanalysator ( 4.9) noch präzise bestimmt werden kann.
Für die Trennung von Threonin-Serin darf das Verhältnis von Tal zur Peakhöhe des niedrigeren der 2 sich überlappenden Peaks auf dem Chromatogramm nicht mehr als 2:10 betragen (wenn nur Cyst(e)in, Methionin, Threonin und Lysin bestimmt werden, beeinträchtigt eine unzureichende Trennung von angrenzenden Peaks die Bestimmung). Für alle übrigen Aminosäuren sollte die Trennung besser als 1:10 sein.
Das System muss gewährleisten, dass Lysin von Lysinartefaktenund Omithin abgetrennt wird.
6. Berechnung der Ergebnisse
Die Flächen der Proben- und der Kalibrationsstandardpeaks werden für jede einzelne Aminosäure bestimmt und der Gehalt (X) in g Aminosäure je kg Probe berechnet:
| X = | a x c x M x V |
| B x m x 1.000 |
| Bei Verwendung eines internen Standards ist zu multiplizieren mit | D C |
a = Peakfläche, Hydrolysat oder Extrakt
B = Peakfläche, Kalibrierstandardlösung
C = Peakfläche, interner Standard im Hydrolysat oder Extrakt
D = Peakfläche, interner Standard, Kalibrierstandardlösung
M = Molmasse der zu bestimmenden Aminosäure
c = Konzentration des Standards inmol/ml
m = Probeneinwaage (auf Originalgewicht berichtigt, falls getrocknet oder entfettet), in g
V = Gesamtvolumen des Hydrolysats ( 5.3.4) in ml oder errechnetes Gesamtverdünnungsvolumen des Extrakts ( 6.1) in ml
Sowohl Cystin als auch Cystein werden im Hydrolysat der oxidierten Probe als Cysteinsäure bestimmt, jedoch als Cystin (C6H12N2O4S2, M = 240,30 g/mol) unter Verwendung der Molmasse von 120,15 g/mol (0,5 × 240,30 g/mol) berechnet.
Methionin wird als Methioninsulfon in Hydrolysaten der oxidierten Probe bestimmt, aber unter Verwendung des M von Methionin (149,21 g/mol) als Methionin berechnet.
Zugesetztes freies Methionin wird nach Extraktion als Methionin bestimmt, wobei für die Berechnung das gleiche M verwendet wird.
6.1 Das Gesamtverdünnungsvolumen der Extrakte (F) für die Bestimmung des Gehalts an freien Aminosäuren ( 5.2) wird wie folgt berechnet:
| F = | 10 ml x (10 ml + 5 ml) | x | V |
| 10 ml | 10 |
V = Volumen des Endextrakts.
7. Beurteilung des Verfahrens
Das Verfahren ist in einem auf internationaler Ebene im Jahr 1990 durchgeführten Vergleichstest an 4 verschiedenen Futtermitteln (Mischfuttermittel für Schweine, Mischfuttermittel für Mastküken, Eiweißkonzentrat und einer Vormischung) erprobt worden. Die nach der Eliminierung von Ausreißern erhaltenen Ergebnisse (Mittelwerte und Standardabweichungen) sind in den Tabellen unter diesem Punkt dargestellt.
Mittelwerte in g/kg
| Referenzmaterial | Aminosäure | |||
| Threonin | Cyst(e)in | Methionin | Lysin | |
| Schweinemischfutter | 6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
| Mastkükenmischfutter | 9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
| Eiweißkonzentrat | 22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
| Vormischung | 58,42 n = 16 |
- | 90,21 n = 16 |
98,03 n = 16 |
| n = Anzahl der beteiligten Laboratorien. | ||||
7.1 Wiederholbarkeit
Für die untersuchten Aminosäuren wurde die Wiederholbarkeit ermittelt. Sie ist, ausgedrückt als Standardabweichung der Wiederholbarkeit, für den oben genannten Vergleichstest in der folgenden Tabelle dargestellt.
Standardabweichung der Wiederholbarkeit (sr) in g/kg
| Referenzmaterial | Aminosäure | |||
| Threonin | Cyst(e)in | Methionin | Lysin | |
| Schweinemischfutter | 0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
| Mastkükenmischfutter | 0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
| Eiweißkonzentrat | 0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
| Vormischung | 1,30 n = 16 |
- | 2,19 n = 16 |
2,06 n = 16 |
| n = Anzahl der beteiligten Laboratorien. | ||||
Variationskoeffizient (%) für die Standardabweichung der Wiederholbarkeit (sr)
| Referenzmaterial | Aminosäure | |||
| Threonin | Cyst(e)in | Methionin | Lysin | |
| Schweinemischfutter | 1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
| Mastkükenmischfutter | 2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
| Eiweißkonzentrat | 2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
| Referenzmaterial | Aminosäure | |||
| Threonin | Cyst(e)in | Methionin | Lysin | |
| Vormischung | 2,2
n = 16 |
- | 2,4
n = 16 |
2,1
n = 16 |
| n = Anzahl der beteiligten Laboratorien. | ||||
7.2 Vergleichbarkeit
Die Ergebnisse hinsichtlich der Standardabweichung der Vergleichbarkeit, die sich aus den oben genannten Untersuchungen ergeben, sind in nachstehender Tabelle dargestellt.
Standardabweichung der Vergleichbarkeit (sR) in g/kg
| Referenzmaterial | Aminosäure | |||
| Threonin | Cyst(e)in | Methionin | Lysin | |
| Schweinemischfutter | 0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
| Mastkükenmischfutter | 0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
| Eiweißkonzentrat | 0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
| Vormischung | 2,49 n = 16 |
- | 6,20 n = 16 |
6,62 n = 16 |
| n = Anzahl der beteiligten Laboratorien. | ||||
Variationskoeffizient (%) für die Standardabweichung der Vergleichbarkeit (sR)
| Referenzmaterial | Aminosäure | |||
| Threonin | Cyst(e)in | Methionin | Lysin | |
| Schweinemischfutter | 4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
| Mastkükenmischfutter | 5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
| Eiweißkonzentrat | 3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
| Vormischung | 4,3 n = 16 |
- | 6,9 n = 16 |
6,7 n = 16 |
| n = Anzahl der beteiligten Laboratorien. | ||||
8. Verwendung von Referenzmaterialien
Die korrekte Anwendung der Methode ist durch Mehrfachbestimmungen an zertifizierten Referenzmaterialien (soweit verfügbar) zu überprüfen. Für die Kalibration wird die Verwendung einer zertifizierten Aminosäurestandardlösung empfohlen.
9. Bemerkungen
9.1 Wegen der Unterschiede zwischen den Aminosäureanalysatoren sind die Endkonzentrationen der Aminosäurenkalibrierlösungen ( 3.27.4 und 3.27.5) und der Hydrolysate ( 5.3.4) als Richtwerte anzusehen.
Der lineare Reaktionsbereich des Geräts muss für alle Aminosäuren geprüft werden.
Die Standardlösung wird mit Citratpufferlösung verdünnt, um Peakflächen in der Mitte des Bereichs zu erhalten.
9.2 Falls Geräte für die Hochleistungsflüssigkeitschromatografie zur Analyse der Hydrolysate eingesetzt werden, sind die Versuchsbedingungen gemäß den Empfehlungen des Herstellers zu optimieren.
9.3 Wird diese Methode für Futtermittel angewandt, die mehr als 1 % Chlorid enthalten (Kraftfuttermittel, Mineralfuttermittel, Ergänzungsfuttermittel), so könnte der Methioningehalt unterschätzt werden, und es ist eine modifizierte Oxidation erforderlich.
G. Bestimmung des Tryptophangehalts
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gesamtgehalts an Tryptophan und des Gehalts an freiem Tryptophan in Futtermitteln. Sie dient nicht zur Unterscheidung zwischen D- und L-Formen.
2. Prinzip
Zur Bestimmung des Gesamtgehalts an Tryptophan wird die Probe unter alkalischen Bedingungen mit gesättigter Bariumhydroxid-Lösung hydrolysiert, auf 110 °C erhitzt und 20 h auf dieser Temperatur gehalten. Nach der Hydrolyse wird ein interner Standard zugegeben.
Zur Bestimmung des Gehalts an freiem Tryptophan wird die Probe unter leicht sauren Bedingungen in Gegenwart eines internen Standards extrahiert.
Tryptophan und der interne Standard im Hydrolysat bzw. im Extrakt werden durch HPLC mit Fluoreszenzdetektor bestimmt.
3. Reagenzien
3.1 Es ist bidestilliertes Wasser oder Wasser gleichwertiger Qualität zu verwenden (Leitfähigkeit < 10 µS/cm).
3.2 Standardsubstanz: Tryptophan (Reinheit/Gehalt ≥ 99 %), im Vakuum über Phosphorpentoxid getrocknet.
3.3 Interner Standard: α-Methyl-Tryptophan (Reinheit/Gehalt ≥ 99 %), im Vakuum über Phosphorpentoxid getrocknet.
3.4 Bariumhydroxid-Octahydrat (das Ba(OH)2.8H2O darf nicht übermäßig der Luft ausgesetzt werden, um die Bildung von BaCO3 zu vermeiden, das die Bestimmung beeinträchtigen könnte) (siehe Bemerkung 9.3).
3.5 Natriumhydroxid.
3.6 Orthophosphorsäure, w (Massenanteil) = 85 %.
3.7 Salzsäure, ρ20 = 1,19 g/ml.
3.8 Methanol, HPLC-Qualität.
3.9 Petrolether, Siedeintervall 40 bis 60 °C.
3.10 Natriumhydroxidlösung, c = 1 mol/l:
40,0 g NaOH (3.5) in Wasser lösen und mit Wasser ( 3.1) auf 1 l auffüllen.
3.11 Salzsäure, c = 6 mol/l:
492 ml HCl ( 3.7) mit Wasser auf 1 l auffüllen.
3.12 Salzsäure, c = 1 mol/l:
82 ml HCl ( 3.7) mit Wasser auf 1 l auffüllen.
3.13 Salzsäure, c = 0,1 mol/l:
8,2 ml HCl ( 3.7) mit Wasser auf 1 l auffüllen.
3.14 Orthophosphorsäure, c = 0,5 mol/l:
34 ml Orthophosphorsäure ( 3.6) mit Wasser ( 3.1) auf 1 l auffüllen.
3.15 Konzentrierte Tryptophanlösung ( 3.2), c = 2,50 µmol/ml:
0,2553 g Tryptophan ( 3.2) in einem 500-ml-Messkolben in Salzsäure ( 3.13) lösen und mit Salzsäure ( 3.13) zur Marke auffüllen. Bei - 18 °C höchstens 4 Wochen aufbewahren.
3.16 Konzentrierte interne Standardlösung, c = 2,50 µmol/ml:
0,2728 g α-Methyl-Tryptophan ( 3.3) in einem 500-ml-Messkolben in Salzsäure ( 3.13) lösen und mit Salzsäure ( 3.13) zur Marke auffüllen. Bei - 18 °C höchstens 4 Wochen aufbewahren.
3.17 Kalibrierstandardlösung von Tryptophan und internem Standard:
2,00 ml konzentrierte Tryptophanlösung ( 3.15) und 2,00 ml konzentrierte interne Standardlösung (α-Methyl-Tryptophan) ( 3.16) mit Wasser ( 3.1) und Methanol ( 3.8) auf etwa dasselbe Volumen und etwa dieselbe Methanolkonzentration (10 bis 30 %) wie das fertige Hydrolysat verdünnen.
Diese Lösung muss vor Gebrauch frisch hergestellt werden.
Während der Zubereitung vor direktem Sonnenlicht schützen.
3.18 Essigsäure.
3.19 1,1,1-Trichlor-2-methyl-2-propanol.
3.20 Ethanolamin, w (Massenanteil) > 98 %.
3.21 Lösung von 1 g 1,1,1-Trichlor-2-methyl-2-propanol ( 3.19) in 100 ml Methanol ( 3.8).
3.22 Mobile Phase für die HPLC: 3,00 g Essigsäure ( 3.18) + 900 ml Wasser (3.1) + 50,0 ml Lösung ( 3.21) von 1,1,1-Trichlor-2-methyl-2-propanol ( 3.19) in Methanol ( 3.8) (1g/100 ml). Der pH-Wert wird mit Ethanolamin ( 3.20) auf 5,00 eingestellt. Mit Wasser ( 3.1) auf 1 l auffüllen.
4. Geräte
4.1 HPLC-Einrichtung mit Fluoreszenzdetektor.
4.2 HPLC-Trennsäule, 125 × 4 mm, C18, 3 µm Korngröße, oder vergleichbare Säule.
4.3 pH-Meter.
4.4 Polypropylen-Kolben, 125 ml, Weithals mit Schraubverschluss.
4.5 Membranfilter, 0,45 µm Porengröße.
4.6 Autoklav, 110 (± 2) °C, 1,4 (± 0,1) bar.
4.7 Mechanischer Schüttler oder Magnetrührer.
4.8 Vortex-Schüttelgerät.
5. Verfahren
5.1 Vorbereitung der Proben
Die Probe wird so fein vermahlen, dass sie ein Sieb mit 0,5 mm Maschenweite passieren kann. Proben mit hohem Feuchtigkeitsgehalt müssen vor dem Vermahlen entweder bei einer Temperatur von höchstens 50 °C luftgetrocknet oder aber gefriergetrocknet werden. Proben mit hohem Fettgehalt sind vor dem Vermahlen mit Petrolether ( 3.9) zu extrahieren.
5.2 Bestimmung des Gehalts an freiem Tryptophan (Extrakt)
Eine geeignete Menge (1 bis 5 g) der vorbereiteten Probe ( 5.1) auf 1 mg genau in einen Erlenmeyerkolben einwiegen. 100,0 ml Salzsäure ( 3.13) und 5,00 ml konzentrierte interne Standardlösung ( 3.16) zugeben. Mit einem mechanischen Schüttler oder einem Magnetrührer ( 4.7) 60 min schütteln bzw. mischen. Absetzen lassen und 10,0 ml der überstehenden Lösung in ein Becherglas pipettieren. 5 ml Orthophosphorsäure ( 3.14) zugeben. Der pH-Wert wird mit Natriumhydroxidlösung ( 3.10) auf 3 eingestellt. Ausreichend Methanol ( 3.8) für eine Methanolkonzentration von 10 bis 30 % im Endvolumen zugeben. In einen Messkolben mit ausreichendem Volumen überführen und mit Wasser auf das für die Chromatografie notwendige Volumen verdünnen (entspricht etwa dem Volumen der Kalibrierstandardlösung ( 3.17)).
Einige ml der Lösung werden vor der Einspritzung in die HPLC-Säule durch einen 0,45-µm-Membranfilter ( 4.5) filtriert. Die Chromatografie gemäß 5.4 durchführen.
Standardlösung und Extrakte sind vor direktem Sonnenlicht zu schützen. Falls es nicht möglich ist, die Extrakte am selben Tag zu analysieren, können sie bei 5 °C maximal 3 Tage aufbewahrt werden.
5.3 Bestimmung des Gehalts an Gesamttryptophan (Hydrolysat)
0,1 bis 1 g der vorbereiteten Probe ( 5.1) werden auf 0,2 mg genau in den Polypropylenkolben ( 4.4) eingewogen. Die Einwaage muss einen Stickstoffgehalt von ca. 10 mg aufweisen. 8,4 g Bariumhydroxid-Octahydrat ( 3.4) und 10 ml Wasser zugeben. Mithilfe eines Vortex-Schüttelgeräts ( 4.8) oder Magnetrührgeräts ( 4.7) mischen. Den teflonbeschichteten Magneten in der Mischung lassen. Die Gefäßwände mit 4 ml Wasser abspülen. Schraubkappe aufsetzen und Kolben locker verschließen. In einem Autoklaven ( 4.6) 30 bis 60 min mit kochendem Wasser erhitzen. Den Autoklaven schließen und 20 h bei 110 (+/2) °C autoklavieren.
Vor dem Öffnen des Autoklaven ist die Temperatur auf knapp unter 100 °C zu senken. Um Kristallisation des Ba(OH)2 × 8HO2 zu vermeiden, sind der warmen Mischung 30 ml Wasser mit Zimmertemperatur zuzugeben. Leicht schütteln oder rühren. 2,00 ml konzentrierte interne Standardlösung (α-Methyl-Tryptophan) ( 3.16) zugeben. Die Gefäße im Wasser-/Eisbad 15 min kühlen.
Anschließend 5 ml Orthophosphorsäure ( 3.14) zugeben. Das Gefäß im Kühlbad belassen und unter Rühren mit HCl ( 3.11) neutralisieren; der pH-Wert wird mit HCl ( 3.12) auf 3,0 eingestellt. Ausreichend Methanol für eine Methanolkonzentration von 10 bis 30 % im Endvolumen zugeben. In einen Messkolben mit ausreichendem Volumen überführen und mit Wasser auf das für die Chromatografie notwendige Volumen verdünnen (z.B. 100 ml). Die Zugabe von Methanol darf keine Präzipitation verursachen.
Einige ml der Lösung werden vor der Einspritzung in die HPLC-Säule durch einen 0,45-µm-Membranfilter ( 4.5) filtriert. Die Chromatografie gemäß 5.4 durchführen.
Standardlösung und Extrakte sind vor direktem Sonnenlicht zu schützen. Falls es nicht möglich ist, die Hydrolysate am selben Tag zu analysieren, können sie bei 5 °C höchstens 3 Tage aufbewahrt werden.
5.4 HPLC-Bestimmung
Die folgenden Angaben für eine isokratische Trennung sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen (siehe auch Bemerkungen 9.1 und 9.2):
| HPLC-Trennsäule (4.2): | 125 × 4 mm, C18, 3 µm Korngröße, oder vergleichbare Säule |
| Säulentemperatur: | Raumtemperatur |
| Mobile Phase (3.22): | 3,00 g Essigsäure ( 3.18) + 900 ml Wasser ( 3.1) + 50,0 ml Lösung (3.21) von 1,1,1-Trichlor-2-methyl-2-propanol ( 3.19) in Methanol ( 3.8) (1 g /100 ml). Den pH-Wert mit Ethanolamin ( 3.20) auf 5,0 einstellen. Mit Wasser ( 3.1) auf 1 l auffüllen. |
| Durchflussrate: | 1 ml/min |
| Gesamtlaufzeit: | ca. 34 min |
| Detektionswellenlänge: | Anregung: 280 nm, Emission: 356 nm |
| Einspritzvolumen: | 20 µl |
6. Berechnung der Ergebnisse
Die Menge von Tryptophan (X) in g je 100 g Probe wird wie folgt berechnet:
a = Peakfläche des internen Standards; Kalibrierstandardlösung ( 3.17)
B = Peakfläche von Tryptophan, Extrakt ( 5.2) oder Hydrolysat (5.3)
V1 = Volumen der konzentrierten Tryptophanlösung ( 3.15), das der Kalibrierlösung ( 3.17) zugegeben wurde, in ml (2 ml)
c = Konzentration der konzentrierten Tryptophanlösung ( 3.15), die der Kalibrierlösung ( 3.17) zugegeben wurde, in µmol/ml (= 2,50)
V2 = Volumen der konzentrierten internen Standardlösung ( 3.16), das bei der Extraktion (5.2) (= 5,00 ml) oder dem Hydrolysat (5.3) (= 2,00 ml) zugegeben wurde, in ml
C = Peakfläche des internen Standards, Extrakt ( 5.2) oder Hydrolysat ( 5.3)
D = Peakfläche von Tryptophan, Kalibrierstandardlösung ( 3.17)
V3 = Volumen der konzentrierten internen Standardlösung (3.16), das der Kalibrierstandardlösung ( 3.17) zugegeben wurde, in ml (2,00 ml)
m = Probeneinwaage (korrigiert auf Originalgewicht, falls getrocknet und/oder entfettet), in g
M = Molmasse von Tryptophan (= 204,23 g/mol)
7. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 10 % des höheren Werts nicht überschreiten.
8. Ergebnisse eines Ringversuchs
Es wurde ein EG-Ringversuch (4. Vergleichstest) durchgeführt, bei dem 3 Proben von bis zu 12 Laboratorien analysiert wurden, um die Hydrolysemethode zu zertifizieren. Jede Probe wurde mehrfach (5-mal) analysiert. Die Ergebnisse sind in nachstehender Tabelle zusammengefasst:
| Probe 1 Schweinefutter |
Probe 2 Schweinefutter mit Zusatz von L-Tryptophan |
Probe 3 Kraftfutter für Schweine |
|
| L | 12 | 12 | 12 |
| n | 50 | 55 | 50 |
| Mittelwert [g/kg] | 2,42 | 3,40 | 4,22 |
| sr[g/kg] | 0,05 | 0,05 | 0,08 |
| r [g/kg] | 0,14 | 0,14 | 0,22 |
| VKr[ %] | 1,9 | 1,6 | 1,9 |
| sR[g/kg] | 0,15 | 0,20 | 0,09 |
| R [g/kg] | 0,42 | 0,56 | 0,25 |
| VKR[ %] | 6,3 | 6,0 | 2,2 |
L = Zahl der Laboratorien, die Ergebnisse übermittelt haben
n = Zahl der Einzelergebnisse ohne Ausreißer (Ausreißertest von Cochran, Dixon)
sr = Standardabweichung der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
r = Wiederholbarkeit
R = Vergleichbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit, in %
VKR = Variationskoeffizient der Vergleichbarkeit, in %
Es wurde ein weiterer EG-Ringversuch (3. Vergleichstest) durchgeführt, bei dem 2 Proben von bis zu 13 Laboratorien analysiert wurden, um die Methode zur Extraktion von freiem Tryptophan zu zertifizieren. Jede Probe wurde mehrfach (5-mal) analysiert. Die Ergebnisse sind in nachstehender Tabelle zusammengefasst:
| Probe 4 Mischung aus Weizen und Soja |
Probe 5 Mischung aus Weizen und Soja (= Probe 4) mit Tryptophan-Zusatz (0,457 g/kg) |
|
| L | 12 | 12 |
| n | 55 | 60 |
| Mittelwert [g/kg] | 0,391 | 0,931 |
| sr[g/kg] | 0,005 | 0,012 |
| r [g/kg] | 0,014 | 0,034 |
| VKr[ %] | 1,34 | 1,34 |
| sR[g/kg] | 0,018 | 0,048 |
| R [g/kg] | 0,050 | 0,134 |
| VKR[ %] | 4,71 | 5,11 |
L = Zahl der Laboratorien, die Ergebnisse übermittelt haben
n = Zahl der Einzelergebnisse ohne Ausreißer (Ausreißertest von Cochran, Dixon)
sr = Standardabweichung der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
r = Wiederholbarkeit
R = Vergleichbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
Es wurde ein weiterer EG-Vergleichstest durchgeführt, bei dem 4 Proben von bis zu 7 Laboratorien analysiert wurden, um die Tryptophan-Hydrolyse zu zertifizieren. Die Ergebnisse sind in nachstehender Tabelle zusammengefasst. Jede Probe wurde mehrfach (5-mal) analysiert.
| Probe 1 Schweine mischfuttermittel (CRM 117) |
Probe 2 Fischmehl mit geringem Fettgehalt (CRM 118) |
Probe 3 Sojamehl (CRM 119) |
Probe 4 Magermilch pulver (CRM 120) |
|
| L | 7 | 7 | 7 | 7 |
| n | 25 | 30 | 30 | 30 |
| Mittelwert [g/kg] | 2,064 | 8,801 | 6,882 | 5,236 |
| s r[g/kg] | 0,021 | 0,101 | 0,089 | 0,040 |
| r [g/kg] | 0,059 | 0,283 | 0,249 | 0,112 |
| VKr[ %] | 1,04 | 1,15 | 1,30 | 0,76 |
| sR[g/kg] | 0,031 | 0,413 | 0,283 | 0,221 |
| R [g/kg] | 0,087 | 1,156 | 0,792 | 0,619 |
| VKR[ %] | 1,48 | 4,69 | 4,11 | 4,22 |
L = Zahl der Laboratorien, die Ergebnisse übermittelt haben
n = Zahl der Einzelergebnisse ohne Ausreißer (Ausreißertest von Cochran, Dixon)
sr = Standardabweichung der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
r = Wiederholbarkeit
R = Vergleichbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
9. Bemerkungen
9.1 Mit den folgenden besonderen Chromatografiebedingungen kann eine bessere Trennung von Tryptophan und α-Methyl-Tryptophan erreicht werden.
Isokratische Trennung, gefolgt von der Reinigung der Gradientensäule:
| HPLC-Trennsäule: | 125 × 4 mm, C18, 5 µm Korngröße, oder vergleichbare Säule | ||
| Säulentemperatur: | 32 °C | ||
| Mobile Phase: | A: 0,01 mol/l KH2PO4/Methanol, 95 + 5 (V+V). B: Methanol |
0 % B | |
| Gradientenprogramm: | 0 min | 100 % A | |
| 15 min | 100 % A | 0 % B | |
| 17 min | 60 % A | 40 % B | |
| 19 min | 60 % A | 40 % B | |
| 21 min | 100 % A | 0 % B | |
| 33 min 100 % A | 0 % B | ||
| Durchflussrate: | 1,2 ml/min | ||
| Gesamtlaufzeit: | ca. 33 min | ||
9.2 Die Durchführung der Chromatografie variiert je nach Art der HPLC und der verwendeten Säulenpackung. Das System muss so gewählt werden, dass eine Grundlinientrennung zwischen Tryptophan und dem internen Standard möglich ist. Die Abbauprodukte müssen deutlich von Tryptophan und dem internen Standard getrennt werden. Zur Untersuchung der Grundlinie unter dem internen Standard auf Verunreinigungen sind Hydrolysate ohne internen Standard zu analysieren. Die Laufzeit muss lang genug sein, dass alle Abbauprodukte eluiert werden, andernfalls können späte Elutionspeaks anschließende Chromatografiedurchgänge beeinträchtigen.
Im Betriebsbereich muss das Chromatografiesystem eine lineare Reaktion ergeben. Die lineare Reaktion ist bei einer konstanten (der normalen) Konzentration des internen Standards und unterschiedlichen Tryptophankonzentrationen zu messen. Die Größe des Tryptophan- und des internen Standardpeaks müssen sich innerhalb des linearen Bereichs des HPLC-/Fluoreszenzsystems befinden. Sollten der Tryptophan- und/oder der interne Standardpeak/die internen Standardpeaks zu klein oder zu hoch sein, ist die Analyse mit einer anderen Probengröße und/oder einem anderen Endvolumen zu wiederholen.
9.3 Bariumhydroxid
Älteres Bariumhydroxid ist schwerer löslich. Dies kann zur Folge haben, dass die Lösung für die HPLCBestimmung nicht klar ist und zu niedrigen Tryptophan-Werten führen kann.
H. Bestimmung des Gehalts an Rohölen und -Fetten
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Rohöl- und Rohfettgehalts von Futtermitteln. Sie erstreckt sich nicht auf die Analyse von Ölsaaten und Ölfrüchten.
Je nach Art und Zusammensetzung des Futtermittels sowie in Abhängigkeit vom Zweck der Untersuchung ist eines der beiden nachstehend beschriebenen Verfahren anzuwenden.
1.1 Verfahren a - Direkt extrahierbare Rohöle und Rohfette
Das Verfahren ist anzuwenden bei Futtermittel-Ausgangserzeugnissen pflanzlicher Herkunft mit Ausnahme derjenigen, die in den Anwendungsbereich von Verfahren B fallen.
1.2 Verfahren B - Gesamtgehalt an Rohölen und Rohfetten
Das Verfahren ist anzuwenden bei Futtermittel-Ausgangserzeugnissen tierischen Ursprungs und bei allen Mischfuttermitteln. Es ist außerdem bei allen Erzeugnissen anzuwenden, aus denen Öle und Fette ohne vorherige Hydrolyse nicht vollständig extrahiert werden können (z.B. Kleber, Hefen, Kartoffeleiweiß und Erzeugnisse, die Verfahren unterzogen wurden wie Extrudieren, Flockieren und Erhitzen).
1.3 Auswertung der Ergebnisse
In allen Fällen, in denen mit Verfahren B ein höheres Ergebnis erzielt wird als mit Verfahren A, gilt das mit Verfahren B erhaltene Ergebnis als der richtige Wert.
2. Prinzip
2.1 Verfahren A
Die Probe wird mit Petrolether extrahiert. Das Lösungsmittel wird abdestilliert und der Rückstand getrocknet und gewogen.
2.2 Verfahren B
Die Probe wird unter Erhitzen mit Salzsäure behandelt. Die Mischung wird abgekühlt und filtriert. Der gewaschene und getrocknete Rückstand wird nach Verfahren a weiterbehandelt.
3. Reagenzien
3.1 Petrolether, Siedeintervall 40 bis 60 °C. Die Bromzahl muss weniger als 1 und der Abdampfrückstand weniger als 2 mg/100 ml betragen.
3.2 Natriumsulfat, wasserfrei.
3.3 Salzsäure, c = 3 mol/l.
3.4 Filterhilfsmittel, z.B. Kieselgur, Hyflo-Supercel.
4. Geräte
4.1 Extraktionsapparat: Sofern das Gerät mit einem Siphon (Soxhletapparat) ausgestattet ist, muss die Rückflussmenge so bemessen sein, dass das Lösungsmittel mindestens 10-mal in der Stunde abläuft. Wenn es sich um ein Gerät ohne Siphon handelt, muss die Rückflussmenge etwa 10 ml/min betragen.
4.2 Extraktionshülsen: Diese müssen frei sein von petroletherlöslichen Stoffen und eine Porosität besitzen, die den Anforderungen nach 4.1 Genüge leistet.
4.3 Trockenschrank: Vakuumtrockenschrank, eingestellt auf 75 °C (± 3 °C,) oder Lufttrockenschrank, eingestellt auf 100 °C (± 3 °C).
5. Verfahren
5.1 Verfahren A (siehe Bemerkung 8.1)
Von der Probe werden 5 g auf 1 mg genau abgewogen, dann in eine Extraktionshülse ( 4.2) überführt und mit einem fettfreien Wattebausch abgedeckt.
Die Hülse wird in einen Extraktionsapparat ( 4.1) eingesetzt und 6 h lang mit Petrolether ( 3.1) extrahiert. Der Petroletherextrakt wird in einem trockenen, mit einigen Körnern Bimsstein1 versehenen, tarierten Kolben aufgefangen.
Nach beendeter Extraktion wird das Lösungsmittel abdestilliert. Anschließend wird der Rückstand mit Kolben 1,5 h im Trockenschrank ( 4.3) getrocknet und nach dem Abkühlen in einem Exsikkator gewogen. Durch eine zweite Trocknung von 30 min Dauer ist zu prüfen, ob das Gewicht der Öle und Fette konstant geblieben ist (der Gewichtsverlust zwischen 2 aufeinanderfolgenden Wägungen darf 1 mg nicht überschreiten).
5.2 Verfahren B
Von der Probe (siehe Bemerkung 8.2) werden 2,5 g auf 1 mg genau abgewogen und in ein 400-ml-Becherglas oder einen 300-ml-Erlenmeyerkolben gebracht. Anschließend werden 100 ml Salzsäure ( 3.3) und einige Körner Bimsstein hinzugefügt. Das Becherglas wird mit einem Uhrglas abgedeckt, bzw. dem Erlenmeyerkolben wird ein Rückflusskühler aufgesetzt. Das Gemisch wird auf kleiner Flamme oder auf einer Heizplatte bis zum Siedepunkt erhitzt und 1 h schwach sieden gelassen. Es ist zu verhindern, dass das Material sich an den Wänden des Gefäßes festsetzt.
Dann wird abgekühlt und so viel vom Filterhilfsmittel ( 3.4) zugesetzt, dass beim Filtrieren kein Öl- bzw. Fettverlust eintritt. Filtriert wird unter Verwendung eines feuchten, fettfreien doppelten Filterpapiers. Der Rückstand wird bis zur neutralen Reaktion mit kaltem Wasser gewaschen. Danach ist zu überprüfen, dass das Filtrat keine Öle und Fette mehr enthält. Bei Vorhandensein von Ölen oder Fetten muss vor der Hydrolyse eine Extraktion der Probe mit Petrolether nach Verfahren a vorgenommen werden.
Das doppelte Filterpapier mit dem Rückstand ist auf ein Uhrglas zu legen und ca. 1,5 h bei 100 °C (± 3 °C) im Lufttrockenschrank ( 4.3) zu trocknen.
Das doppelte Filterpapier mit dem trockenen Rückstand wird in eine Extraktionshülse ( 4.2) überführt und mit einem fettfreien Wattebausch abgedeckt. Die Hülse wird in einen Extraktionsapparat ( 4.1) eingesetzt. Dann wird, wie unter 5.1 Absätze 2 und 3 beschrieben, weiterverfahren.
6. Berechnung der Ergebnisse
Das Gewicht des Rückstands wird als Prozentsatz der Probe ausgedrückt.
7. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen eines Analytikers darf bei ein und derselben Probe die folgenden Werte nicht überschreiten:
8. Bemerkungen
8.1 Bei Erzeugnissen mit hohem Öl- und Fettgehalt, die schwer zu zerkleinern sind oder sich zur Entnahme einer reduzierten homogenen Probe nicht eignen, ist folgendermaßen zu verfahren:
Von der Probe werden 20 g auf 1 mg genau abgewogen und mit 10 g oder mehr wasserfreiem Natriumsulfat ( 3.2) gemischt. Dann wird mit Petrolether ( 3.1), wie unter 5.1 beschrieben, extrahiert. Der dabei aufgefangene Extrakt wird mit Petrolether ( 3.1) auf 500 ml aufgefüllt und gemischt. Von der Lösung werden 50 ml entnommen und in einen kleinen, trockenen, mit Bimssteinkörnchen versehenen und tarierten Kolben gebracht. Das Lösungsmittel wird abdestilliert und dann wird, wie unter 5.1 letzter Absatz beschrieben, getrocknet und weiterverfahren.
Der Extraktionsrückstand in der Hülse wird vom Lösungsmittel befreit, auf 1 mm zerkleinert und wieder zurück in die Extraktionshülse gebracht (ohne Zugabe von Natriumsulfat). Dann wird, wie unter 5.1 Absätze 2 und 3 beschrieben, fortgefahren.
Der Gehalt an Ölen und Fetten, ausgedrückt als Prozentsatz der Probe, wird nach folgender Formel berechnet:
(10 m1+ m2) × 5
wobei:
m1 = Gewicht des Rückstands in g nach der ersten Extraktion (aliquoter Teil des Extrakts),
m2 = Gewicht des Rückstands in g nach der zweiten Extraktion.
8.2 Bei öl- und fettarmen Erzeugnissen kann mit einer Einwaage von 5 g gearbeitet werden.
8.3 Heimtierfutter mit hohem Wassergehalt muss vor der Hydrolyse und Extraktion nach Verfahren B möglicherweise mit wasserfreiem Natriumsulfat gemischt werden.
8.4 Bei dem Verfahren nach 5.2 kann die Verwendung von heißem statt kaltem Wasser zum Waschen des Rückstands nach der Filtration möglicherweise wirksamer sein.
8.5 Die Trocknungszeit von 1,5 h muss bei einigen Futtermitteln möglicherweise verlängert werden. Übermäßiges Trocknen ist zu vermeiden, da dies zu niedrigen Ergebnissen führen kann. Es kann auch ein Mikrowellengerät verwendet werden.
8.6 Bei einem Rohöl/-fettgehalt von mehr als 15 % wird vor der Hydrolyse eine Extraktion nach Verfahren a und danach eine erneute Extraktion nach Verfahren B empfohlen. Dies hängt zum Teil von der Art des Futtermittels und der Art des Rohfetts im Futtermittel ab.
_________
1) Soll das Öl oder Fett später einer Qualitätsprüfung unterzogen werden, sind die Bimssteinkörner durch Glasperlen zu ersetzen.
I. Bestimmung des Rohfasergehalts
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an säure- und alkaliunlöslichen, fettfreien organischen Bestandteilen in Futtermitteln, herkömmlicherweise als Rohfaser bezeichnet.
2. Prinzip
Die gegebenenfalls entfettete Probe wird nacheinander mit kochender Schwefelsäurelösung und kochender Kaliumhydroxidlösung definierter Konzentrationen behandelt. Der Rückstand wird durch Filtration auf einem gesinterten Glasfilter getrennt, gewaschen, getrocknet, gewogen und bei 475 bis 500 °C verascht. Der Gewichtsverlust bei dieser Veraschung entspricht der Rohfaser der Einwaage.
3. Reagenzien
3.1 Schwefelsäure, c = 0,13 mol/l.
3.2 Antischaummittel (z.B. n-Oktanol).
3.3 Filterhilfsmittel (Celite 545 oder gleichwertiges Mittel), 4 h auf 500 °C erhitzt ( 8.6).
3.4 Aceton.
3.5 Petrolether, Siedeintervall 40 bis 60 °C.
3.6 Salzsäure, c = 0,5 mol/l.
3.7 Kaliumhydroxidlösung, c = 0,23 mol/l.
4. Geräte
4.1 Heizvorrichtung für den Aufschluss mit Schwefelsäure und Kaliumhydroxidlösung, ausgestattet mit einer Halterung für den Glasfiltertiegel ( 4.2) und einem Abflussrohr mit Hahn zum Flüssigkeitsablauf, für den Vakuumanschluss und, sofern möglich, auch mit einem Anschluss für die Zufuhr von Druckluft. Vor der täglichen Inbetriebnahme muss die gesamte Apparatur mit Wasser 5 min erhitzt werden.
4.2 Glasfiltertiegel mit eingeschmolzenem gesintertem Glasfilter (Porengröße 40 bis 90 μm). Vor dem erstmaligen Gebrauch wird der Tiegel einige Minuten bei 500 °C geglüht und dann abkühlen gelassen ( 8.6).
4.3 Siedezylinder (mindestens 270 ml) mit einem Rückflusskühler.
4.4 Trockenschrank mit Thermostat.
4.5 Muffelofen mit Thermostat.
4.6 Extraktionsvorrichtung mit einer Halterung für den Glasfiltertiegel ( 4.2) und einem Abflussrohr mit Hahn für den Flüssigkeitsablauf und den Vakuumanschluss.
4.7 Verbindungsringe zur Verbindung von Heizvorrichtung ( 4.1), Filtertiegel ( 4.2) und Siedezylinder ( 4.3), Verbindungsringe zur Verbindung von Kaltextraktionsvorrichtung ( 4.6) und Filtertiegel.
5. Verfahren
Von der vorbereiteten Probe werden 1 g auf 1 mg genau in den Glasfiltertiegel ( 4.2) eingewogen (siehe Bemerkungen 8.1, 8.2 und 8.3), und es wird 1 g Filterhilfsmittel ( 3.3) hinzugefügt.
Der Glasfiltertiegel ( 4.2) wird in die Heizvorrichtung ( 4.1) eingesetzt und mit dem Siedezylinder ( 4.3) verbunden. 150 ml auf Siedetemperatur erhitzte Schwefelsäure ( 3.1) werden in den mit dem Tiegel verbundenen Siedezylinder überführt, und es werden erforderlichenfalls einige Tropfen Antischaummittel ( 3.2) hinzugefügt.
Die Flüssigkeit wird innerhalb von 5 ± 2 min zum Sieden erhitzt und genau 30 min im kräftigen Sieden gehalten.
Der Hahn im Abflussrohr ( 4.1) wird geöffnet und die Schwefelsäure mithilfe von Vakuum durch den Glasfiltertiegel abgesaugt. Der Filterrückstand wird 3-mal mit je 30 ml kochendem Wasser gewaschen. Nach jedem Waschvorgang ist der Rückstand trocken zu saugen.
Der Auslaufhahn wird geschlossen, und es werden 150 ml siedende Kaliumhydroxidlösung ( 3.7) und einige Tropfen Antischaummittel ( 3.2) in den mit dem Siedezylinder verbundenen Glasfiltertiegel gegeben. Die Flüssigkeit wird innerhalb von 5 ± 2 min zum Sieden erhitzt und genau 30 min im kräftigen Sieden gehalten. Die Filtration und das Waschen des Rückstands werden in der gleichen Weise durchgeführt wie nach der Behandlung mit Schwefelsäure.
Nach dem letzten Waschen wird der Rückstand trocken gesaugt und der Tiegel mit Inhalt vom Siedezylinder gelöst und an die Kaltextraktionsvorrichtung ( 4.6) angeschlossen. Unter Anlegen von Vakuum wird der Rückstand 3-mal mit je 25 ml Aceton ( 3.4) gewaschen, wobei der Rückstand nach jedem Waschvorgang trocken zu saugen ist.
Der Filtertiegel mit Inhalt wird im Trockenschrank bei 130 °C bis zur Massekonstanz getrocknet, in einem Exsikkator abgekühlt und anschließend rasch gewogen. Danach wird der Filtertiegel mit Inhalt in einen Muffelofen gebracht und mindestens 30 min bei einer Temperatur von 475 bis 500 °C bis zur Massekonstanz verascht (der Gewichtsverlust in 2 aufeinanderfolgenden Wägungen darf 2 mg nicht überschreiten).
Nach dem Erhitzen wird der Tiegel zunächst im Muffelofen und dann im Exsikkator abgekühlt und anschließend gewogen.
Es wird ein Blindversuch ohne Probe durchgeführt. Der beim Veraschen auftretende Gewichtsverlust darf 4 mg nicht überschreiten.
6. Berechnung der Ergebnisse
Der Gehalt an Rohfaser als Prozentsatz der Probe wird nach folgender Formel berechnet: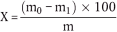
wobei:
m = Einwaage in g,
m0 = Gewichtsverlust nach dem Veraschen während der Bestimmung, in g,
m1 = Gewichtsverlust nach dem Veraschen während des Blindversuchs, in g.
7. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
8. Bemerkungen
8.1 Futtermittel, welche mehr als 10 % Rohfett enthalten, müssen vor der Bestimmung mit Petrolether ( 3.5) entfettet werden. Dazu wird der Glasfiltertiegel ( 4.2) mit Inhalt an die Kaltextraktionsvorrichtung ( 4.6) angeschlossen und unter Anlegen von Vakuum 3-mal mit je 30 ml Petrolether gewaschen, wobei sichergestellt wird, dass der Rückstand trocken ist. Der Tiegel mit Inhalt wird an die Heizvorrichtung ( 4.1) angeschlossen. Danach ist gemäß 5 fortzufahren.
8.2 Futtermittel, die Fette enthalten, welche nicht direkt mit Petrolether ( 3.5) extrahiert werden können, müssen, wie unter 8.1 angegeben, entfettet werden und nach dem Sieden mit Säure ein weiteres Mal entfettet werden. Nach dem Sieden mit Säure und dem anschließenden Waschvorgang wird der Tiegel mit Inhalt an die Kaltextraktionsvorrichtung ( 4.6) angeschlossen und 3-mal mit je 30 ml Aceton und danach weitere 3-mal mit je 30 ml Petrolether entfettet. Der Filtertiegel ( 4.2) wird trocken gesaugt und die Analyse, wie unter 5 beschrieben, fortgesetzt, beginnend mit der Behandlung mit Kaliumhydroxidlösung.
8.3 Bei Futtermitteln, die mehr als 5 % Carbonate, ausgedrückt als Calciumcarbonat, enthalten, wird der Tiegel ( 4.2) mit der eingewogenen Probemenge an die Heizvorrichtung ( 4.1) angeschlossen. Die Probe wird 3-mal mit je 30 ml Salzsäure ( 3.6) gewaschen. Nach jeder Zugabe wird die Probe vor dem Absaugen etwa 1 min stehen gelassen. Anschließend wird 1-mal mit 30 ml Wasser gewaschen. Danach ist, wie unter 5 angegeben, fortzufahren.
8.4 Wenn ein Gerät benutzt wird, bei dem mehrere Glasfiltertiegel an derselben Heizvorrichtung gleichzeitig befestigt sind, dürfen 2 Einzelbestimmungen derselben Probe nicht in derselben Untersuchungsreihe vorgenommen werden.
8.5 Wenn nach dem Sieden mit Säure bzw. Lauge Schwierigkeiten bei der Filtration eintreten, wird Druckluft durch das Auslassrohr der Heizvorrichtung zugeführt und anschließend die Filtration fortgesetzt.
8.6 Die Veraschungstemperatur darf 500 °C nicht überschreiten, um die Haltbarkeit der Glasfiltertiegel zu verlängern. Beim Erhitzen und Abkühlen sind vor allem übermäßige Temperatursprünge zu vermeiden.
J. Bestimmung des Zuckergehalts
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an reduzierenden Zuckern und des Gesamtzuckers nach Inversion, ausgedrückt als Glucose oder gegebenenfalls nach Multiplikation mit dem Faktor 0,95 als Saccharose. Sie wird bei Mischfuttermitteln angewendet. Für andere Futtermittel sind besondere Verfahren vorgesehen. Gegebenenfalls muss der Gehalt an Lactose getrennt bestimmt und dieses Ergebnis bei der Berechnung berücksichtigt werden.
2. Prinzip
Die Zucker werden in verdünntem Ethanol gelöst; die Lösung wird mit den Lösungen Carrez I und II geklärt. Nach dem Verdunsten des Ethanols werden vor und nach der Inversion die Bestimmungen nach der Luff-Schoorl-Methode durchgeführt.
3. Reagenzien
3.1 Ethanollösung (Volumenkonzentration = 40 %), Dichte: 0,948 g/ml bei 20 °C, auf den Umschlagspunkt von Phenolphthalein eingestellt.
3.2 Carrez-Lösung I: 21,9 g Zinkacetat, Zn(CH3 COO)2⋅ 2H2O, und 3 g Eisessig werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt.
3.3 Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat (II), K4 Fe(CN)6⋅ 3H2O, werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt.
3.4 Methylorangelösung (Massenkonzentration = 0,1 %).
3.5 Salzsäure 4 mol/l.
3.6 Salzsäure 0,1 mol/l.
3.7 Natriumhydroxidlösung 0,1 mol/l.
3.8 Reagenz nach Luff-Schoorl:
Die Zitronensäurelösung ( 3.8.2) ist unter vorsichtigem Rühren in die Natriumcarbonatlösung ( 3.8.3) zu gießen. Nach Zugabe der Kupfersulfatlösung ( 3.8.1) ist mit Wasser auf 1 l aufzufüllen. Über Nacht absetzen lassen und dann filtrieren.
Die Konzentration des so erhaltenen Reagenz (Cu 0,05 mol/l; Na2 CO31 mol/l) muss geprüft werden, siehe 5.4 letzter Absatz. Der pH-Wert der Lösung muss bei etwa 9,4 liegen.
3.8.1 Kupfersulfatlösung: 25 g Kupfersulfat, CuSO4 ⋅ 5H2O, eisenfrei, werden in 100 ml Wasser gelöst.
3.8.2 Zitronensäurelösung: 50 g Zitronensäure, C6H8O7⋅ H2O, werden in 50 ml Wasser gelöst.
3.8.3 Natriumcarbonatlösung: 143,8 g Natriumcarbonat, wasserfrei, werden in ca. 300 ml warmem Wasser gelöst. Die Lösung wird abkühlen gelassen.
3.9 Natriumthiosulfatlösung 0,1 mol/l.
3.10 Stärkelösung: eine Aufschlämmung von 5 g löslicher Stärke in 30 ml Wasser wird zu 1 l siedendem Wasser hinzugefügt und 3 min lang im Sieden gehalten; dann wird abkühlen gelassen und gegebenenfalls 10 mg Quecksilberiodid als Konservierungsmittel hinzugefügt.
3.11 Schwefelsäure 3 mol/l.
3.12 Kaliumiodidlösung (Massenkonzentration = 30 %).
3.13 Bimssteinkörner, mit Salzsäure ausgekocht, mit Wasser gewaschen und getrocknet.
3.14 3-Methylbutan-1-ol.
4. Geräte
Mechanisches Schüttelgerät, ca. 35 bis 40 min-1.
5. Verfahren
5.1 Extraktion der Probe
Von der Probe werden 2,5 g auf 1 mg genau in einen 250-ml-Messkolben eingewogen. Nach Zugabe von 200 ml Ethanol ( 3.1) wird der Kolben 1 h lang im Schüttelgerät gemischt. Anschließend werden 5 ml Carrez-Lösung I ( 3.2) hinzugefügt, und es wird ca. 30 s lang gerührt. Dann werden 5 ml Carrez-Lösung II ( 3.3) hinzugefügt und 1 min lang gerührt; nun wird mit Ethanol ( 3.1) zur Marke aufgefüllt, homogenisiert und filtriert. Vom Filtrat werden 200 ml abpipettiert und auf ungefähr die Hälfte des Volumens eingeengt, um den größten Teil des Ethanols zur Verdunstung zu bringen. Der Abdampfrückstand wird mit warmem Wasser in einen 200-ml-Messkolben übergespült, abgekühlt, mit Wasser zur Marke aufgefüllt, homogenisiert und, falls erforderlich, filtriert. Diese Lösung wird für die Bestimmung des Gehalts an reduzierenden Zuckern und nach Inversion zur Bestimmung des Gesamtzuckers verwendet.
5.2 Bestimmung des Gehalts an reduzierenden Zuckern
Eine Menge von höchstens 25 ml der Lösung, die weniger als 60 mg reduzierende Zucker, ausgedrückt als Glucose, enthält, wird abpipettiert, falls erforderlich mit destilliertem Wasser auf 25 ml aufgefüllt und der Gehalt an reduzierendem Zucker nach der Luff-Schoorl-Methode bestimmt. Das Ergebnis wird als Prozentsatz Glucose in der Probe ausgedrückt.
5.3 Bestimmung des Gehalts an Gesamtzucker nach Inversion
Von der Lösung werden 50 ml in einen 100-ml-Messkolben pipettiert, einige Tropfen Methylorangelösung ( 3.4) hinzugefügt und dann vorsichtig unter ständigem Rühren Salzsäure ( 3.5) bis zum eindeutigen Umschlag nach Rot hinzugegeben. Dann werden 15 ml Salzsäure ( 3.6) hinzugefügt, und der Kolben wird 30 min lang in ein Bad mit kräftig siedendem Wasser gestellt, dann schnell auf die Temperatur von etwa 20 °C abgekühlt und 15 ml Natriumhydroxidlösung ( 3.7) hinzugegeben. Der Kolben wird mit Wasser auf 100 ml aufgefüllt, geschüttelt und eine Menge von höchstens 25 ml entnommen, die weniger als 60 mg reduzierende Zucker, ausgedrückt als Glucose, enthält. Falls erforderlich, wird mit destilliertem Wasser auf 25 ml aufgefüllt und der Gehalt an reduzierenden Zuckern nach der Luff-Schoorl-Methode bestimmt. Das Ergebnis wird als Prozentsatz Glucose oder gegebenenfalls nach Multiplikation mit dem Faktor 0,95 als Saccharose ausgedrückt.
5.4 Titration nach Luff-Schoorl
In einen 300-ml-Erlenmeyerkolben werden 25 ml Reagenz nach Luff-Schoorl ( 3.8) pipettiert und anschließend genau 25 ml der geklärten Zuckerlösung hinzugefügt. Nach Zugabe von 2 Körnchen Bimsstein ( 3.13) wird unter Schütteln mit der Hand über freier Flamme von mittlerer Höhe erhitzt, so dass die Flüssigkeit in etwa 2 min zum Sieden kommt. Anschließend wird der Erlenmeyerkolben sofort auf ein Drahtnetz mit einer Asbestscheibe, in die ein Loch von etwa 6 cm Durchmesser gestanzt wurde, gestellt; vorher wird unter dem Drahtnetz eine Flamme entzündet und so eingestellt, dass der Kolben nur am Boden erhitzt wird. Dann wird der Kolben mit einem Rückflusskühler verbunden. Von diesem Augenblick an wird der Kolben genau 10 min sieden gelassen und anschließend sofort in kaltem Wasser abgekühlt. Nach etwa 5 min wird wie folgt titriert:
Der Flüssigkeit werden 10 ml Kaliumiodidlösung ( 3.12) und unmittelbar danach, aber vorsichtig (wegen der Gefahr des übermäßigen Schäumens), 25 ml Schwefelsäure ( 3.11) zugegeben. Dann wird mit Natriumthiosulfatlösung (lc 3.9) zunächst bis zum Auftreten einer mattgelben Farbe titriert und nach Zugabe von Stärkelösung ( 3.10) als Indikator die Titration zu Ende geführt.
Die gleiche Titration wird in einer Mischung aus genau 25 ml Reagenz nach Luff-Schoorl ( 3.8) und 25 ml Wasser nach Zugabe von 10 ml Kaliumiodidlösung ( 3.12) und 25 ml Schwefelsäure ( 3.11) durchgeführt, jedoch ohne vorheriges Erhitzen.
6. Berechnung der Ergebnisse
Aus der unten angeführten Tabelle wird die Menge Glucose in mg abgelesen, die der Differenz der Ergebnisse beider Titrationen, ausgedrückt in ml Natriumthiosulfatlösung (0,1 mol/l), entspricht. Das Ergebnis wird als Prozentsatz der Probe angegeben.
7. Sonderverfahren
7.1 Bei sehr stark melassehaltigen Futtermitteln und anderen wenig homogenen Futtermitteln werden 20 g in einen 1-l-Messkolben eingewogen, 500 ml Wasser hinzugefügt und 1 h lang im Schüttelgerät gemischt. Danach wird - jedoch mit jeweils der 4-fachen Menge der Carrez-Lösungen I ( 3.2) und II ( 3.3) - wie unter 5.1 beschrieben geklärt. Anschließend wird mit Ethanol (Volumenkonzentration = 80 %) zur Marke aufgefüllt.
Die Lösung wird homogenisiert und filtriert. Das Ethanol wird, wie unter 5.1 beschrieben, entfernt. Bei Abwesenheit verkleisterter Stärke wird mit Wasser zur Marke aufgefüllt.
7.2 Bei Melasse und Futtermittel-Ausgangserzeugnissen, die reich an Zucker, aber praktisch stärkefrei sind (Johannisbrot, Zuckerrübentrockenschnitzel usw.), werden 5 g in einen 250-ml-Messkolben eingewogen und 200 ml destilliertes Wasser hinzugefügt. Dann wird 1 h lang oder, falls erforderlich, länger im Schüttelgerät gemischt. Danach wird mit den Carrez-Lösungen I (3.2) und II ( 3.3), wie unter 5.1 beschrieben, geklärt und anschließend mit Wasser zur Marke aufgefüllt, homogenisiert und filtriert. Zur Bestimmung des Gesamtzuckergehalts wird, wie unter 5.3 beschrieben, verfahren.
8. Bemerkungen
8.1 Es wird empfohlen, vor dem Erhitzen mit dem Luff-Schoorl-Reagenz (unabhängig vom Volumen) etwa 1 ml 3-Methylbutan-1-ol ( 3.14) hinzuzufügen, um die Schaumbildung zu vermeiden.
8.2 Die Differenz zwischen dem Gehalt an Gesamtzucker nach Inversion, ausgedrückt als Glucose, und dem Gehalt an reduzierenden Zuckern, ausgedrückt als Glucose, ergibt nach Multiplikation mit dem Faktor 0,95 das Ergebnis als Prozentsatz Saccharose.
8.3 Zur Bestimmung des Gehalts an reduzierenden Zuckern, mit Ausnahme von Milchzucker (Lactose), gibt es 2 Möglichkeiten:
8.3.1 Für eine annähernde Berechnung wird der aus einer getrennten Bestimmung ermittelte Lactosegehalt mit 0,675 multipliziert und das Ergebnis von dem Gehalt an reduzierenden Zuckern abgezogen.
8.3.2 Für eine genaue Berechnung der reduzierenden Zucker (mit Ausnahme von Lactose) ist es notwendig, dass beiden endgültigen Bestimmungen die gleiche Menge der Einwaage zugrunde liegt. Die eine Bestimmung wird in einem Teil der nach 5.1. hergestellten Lösung durchgeführt, die zweite Bestimmung in einem Teil der Lösung, die bei der Bestimmung des Lactosegehalts mittels der für diesen Zweck vorgesehenen Methode (nach Vergärung der anderen Zuckerarten und Klärung) hergestellt wird.
In beiden Fällen wird der anwesende Zucker nach der Luff-Schoorl-Methode bestimmt und in mg Glucose berechnet. Diese beiden Werte werden voneinander subtrahiert und die Differenz wird als Prozentsatz der Probe ausgedrückt.
Beispiel:
Die beiden abpipettierten Mengen entsprechen bei jeder Bestimmung einer Probeneinwaage von 250 mg.
Im ersten Fall werden 17 ml Natriumthiosulfatlösung 0,1 mol/l, entsprechend 44,2 mg Glucose, verbraucht; im zweiten Fall werden 11 ml, entsprechend 27,6 mg Glucose, verbraucht.
Die Differenz beträgt also 16,6 mg Glucose.
Der Gehalt an reduzierenden Zuckern (mit Ausnahme von Lactose), berechnet als Glucose, ist demnach
| 4 x 16,6 | = 6,64 % |
| 10 |
Tabelle für 25 ml Reagenz nach Luff-Schoorl
ml Na2 S2 O30,1 mol/l, 2 min erhitzen, 10 min sieden
| Na2 S2 O3 0,1 mol/l |
Glucose, Fructose, Invertzucker C6 H12 O6 | Lactose C12 H22 O11 |
Maltose C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
| ml | mg | Differenz | mg | Differenz | Mg | Differenz | ml |
| 1 | 2,4 | 2,4 | 3,6 | 3,7 | 3,9 | 3,9 | 1 |
| 2 | 4,8 | 2,4 | 7,3 | 3,7 | 7,8 | 3,9 | 2 |
| 3 | 7,2 | 2,5 | 11,0 | 3,7 | 11,7 | 3,9 | 3 |
| 4 | 9,7 | 2,5 | 14,7 | 3,7 | 15,6 | 4,0 | 4 |
| 5 | 12,2 | 2,5 | 18,4 | 3,7 | 19,6 | 3,9 | 5 |
| 6 | 14,7 | 2,5 | 22,1 | 3,7 | 23,5 | 4,0 | 6 |
| 7 | 17,2 | 2,6 | 25,8 | 3,7 | 27,5 | 4,0 | 7 |
| 8 | 19,8 | 2,6 | 29,5 | 3,7 | 31,5 | 4,0 | 8 |
| 9 | 22,4 | 2,6 | 33,2 | 3,8 | 35,5 | 4,0 | 9 |
| 10 | 25,0 | 2,6 | 37,0 | 3,8 | 39,5 | 4,0 | 10 |
| 11 | 27,6 | 2,7 | 40,8 | 3,8 | 43,5 | 4,0 | 11 |
| 12 | 30,3 | 2,7 | 44,6 | 3,8 | 47,5 | 4,1 | 12 |
| 13 | 33,0 | 2,7 | 48,4 | 3,8 | 51,6 | 4,1 | 13 |
| 14 | 35,7 | 2,8 | 52,2 | 3,8 | 55,7 | 4,1 | 14 |
| 15 | 38,5 | 2,8 | 56,0 | 3,9 | 59,8 | 4,1 | 15 |
| 16 | 41,3 | 2,9 | 59,9 | 3,9 | 63,9 | 4,1 | 16 |
| 17 | 44,2 | 2,9 | 63,8 | 3,9 | 68,0 | 4,2 | 17 |
| 18 | 47,1 | 2,9 | 67,7 | 4,0 | 72,2 | 4,3 | 18 |
| 19 | 50,0 | 3,0 | 71,7 | 4,0 | 76,5 | 4,4 | 19 |
| 20 | 53,0 | 3,0 | 75,7 | 4,1 | 80,9 | 4,5 | 20 |
| 21 | 56,0 | 3,1 | 79,8 | 4,1 | 85,4 | 4,6 | 21 |
| 22 | 59,1 | 3,1 | 83,9 | 4,1 | 90,0 | 4,6 | 22 |
| 23 | 62,2 | 88,0 | 94,6 | 23 | |||
|
weiter . | |
(Stand: 19.03.2024)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: 90.- € netto (Grundlizenz)
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion