Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 1976, Lebensmittel - Futtermittel

| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 1976, Lebensmittel - Futtermittel |
|
Siebte Richtlinie 76/372/EWG der Kommission vom 1. März 1976 zur Festlegung gemeinschaftlicher Analysemethoden für die amtliche Untersuchung von Futtermitteln
(ABl. Nr. L 102 vom 15.04.1976 S. 8;
RL 81/680/EWG - ABl. Nr. L 246 vom 29.08.1981 S. 3;
RL 92/95/EWG - ABl. Nr. L 327 vom 13.11.1992 S. 54;
RL 94/14/EG - ABl. Nr. L 94 vom 13.04.1994 S. 30;
VO (EG) 152/2009 - ABl. Nr. L 54 vom 26.02.2009 S. 1aufgehoben)
aufgehoben/ersetztgem. Art. 6 der VO(EG) 152/2009 - Entsprechungstabelle
Die Kommission der Europäischen Gemeinschaften -
gestützt auf den Vertrag zur Gründung der Europäischen Wirtschaftsgemeinschaft,
gestützt auf die Richtlinie des Rates vom 20. Juli 1970 über die Einführung gemeinschaftlicher Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln 1, zuletzt geändert durch die Beitrittsakte 2, insbesondere auf Artikel 2,
in Erwägung nachstehender Gründe:
Die obengenannte Richtlinie bestimmt, dass die amtlichen Untersuchungen von Futtermitteln zur Feststellung, ob die auf Grund der Rechts- oder Verwaltungsvorschriften festgelegten Anforderungen hinsichtlich Beschaffenheit und Zusammensetzung der Futtermittel erfüllt sind, nach gemeinschaftlichen Probenahmeverfahren und Analysemethoden durchgeführt werden.
Die Richtlinien 71/250/EWG, 71/393/EWG, 72/199/EWG, 73/46/EWG, 74/203/EWG und 75/84/EWG der Kommission vom 15. Juni 1971 3, vom 18. November 1971 4, vom 27. April 1972 5, vom 5. Dezember 1972 6, vom 25. März 1974 7 und vom 20. Dezember 1974 8 haben bereits eine Reihe von Analysemethoden festgelegt. Der Stand der seitdem durchgeführten Arbeiten ermöglicht es nunmehr, eine siebente Reihe von Analysemethoden festzulegen.
Die in dieser Richtlinie vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Futtermittelausschusses
- hat folgende Richtlinie erlassen:
Die Mitgliedstaaten schreiben vor, dass die Analysen für die amtliche Untersuchung von Futtermitteln auf ihren Gehalt an Aflatoxin B1 nach den in der Anlage zu dieser Richtlinie beschriebenen Methoden durchgeführt werden.
Die Mitgliedstaaten setzen spätestens zum 1. Oktober 1976 die erforderlichen Rechts- oder Verwaltungsvorschriften in Kraft, um den Bestimmungen dieser Richtlinie nachzukommen. Sie setzen die Kommission unverzüglich hiervon in Kenntnis.
Diese Richtlinie ist an alle Mitgliedstaaten gerichtet.
| Bestimmung von Aflatoxin B1 | Anlage |
A. Methode mit eindimensionaler Dünnschichtchromatographie
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an Aflatoxin B1 in Rohstoffen und Einzelfuttermitteln. Diese Methode kann nicht angewendet werden, wenn Citrustrester vorhanden ist. Die untere Grenze der Bestimmbarkeit beträgt 0,01 mg/kg (10 ppb).
Wenn Störsubstanzen vorhanden sind, welche die Auswertung erschweren, muss die Analyse nach der Methode B (Hochleistungsflüssigkeitschromatographie) wiederholt werden.
2. Prinzip
Die Probe wird mit Chloroform extrahiert. Der Extrakt wird filtriert und ein aliquoter Teil durch Chromatographie an einer Kieselgelsäule gereinigt. Das Eluat wird eingedampft und der Rückstand in einer definierten Menge Chloroform oder Benzol-Acetonitrilgemisch gelöst. Ein aliquoter Teil dieser Lösung wird dünnschichtchromatographisch untersucht. Die Aflatoxin-B1-Menge wird im UV-Licht visuell oder fluorodensitometrisch im Vergleich zu bekannten Mengen Aflatoxin-B1-Standard ermittelt. Die Identität von Aflatoxin B1 im Futtermittelextrakt muss durch das angegebene Verfahren bestätigt werden.
3. Reagenzien
Anmerkung: Wenn nichts anderes angegeben ist, sind Reagenzien von p.a."-Qualität zu verwenden.
3.1. Aceton
3.2. Chloroform, mit 0,5 bis 1,0 % Äthanol 96 % ig (V/V) stabilisiert
3.3. n-Hexan
3.4. Methanol
3.5. Diäthyläther, wasserfrei, peroxidfrei
3.6. Benzol-Acetonitril-Gemisch 98:2 (V/V)
3.7. Mischung aus Chloroform ( 3.2) und Methanol (3.4) 97:3 (V/V)
3.8. Kieselgel für Säulenchromatographie, 0,05 bis 0,20 mm Teilchengröße
3.9. Watte, hydrophil und mit Chloroform entfettet, oder Glaswolle
3.10. Natriumsulfat, wasserfrei, gekörnt
3.11. Inertgas, z.B. Stickstoff
3.12. 1 N-Salzsäure
3.13. Schwefelsäure 50 % (V/V)
3.14. Kieselgur (Hyflosupercel), mit Säure gewaschen
3.15. Kieselgel G-HR oder gleichwertiges für Dünnschichtchromatographie
3.16. Standardlösung mit etwa 0,1 µg Aflatoxin B1je ml in Chloroform ( 3.2) oder in Benzol-Acetonitril-Gemisch ( 3.6), hergestellt und kontrolliert nach Punkt 7.
3.17. Qualitative Standardlösung mit etwa 0,1 µg Aflatoxin B1 und B2 je ml in Chloroform ( 3.2) oder Benzol-Acetonitril-Gemisch ( 3.6). Diese Konzentrationen sind als Anhaltspunkte gegeben. Sie sind, entsprechend dem unterschiedlichen Fluoreszenzvermögen, so zu wählen, dass sich für beide Aflatoxine dieselbe Fluoreszenzintensität ergibt.
3.18. Fließmittel:
3.18.1. Mischung aus Chloroform ( 3.2) und Aceton ( 3.1) 9:1 (V/V). Trennkammer mit nicht gesättigter Atmosphäre
3.18.2. Mischung aus Diäthyläther ( 3.5), Methanol ( 3.4) und Wasser 96:3:1 (V/V/V). Trennkammer mit nicht gesättigter Atmosphäre
3.18.3. Mischung aus Diäthyläther ( 3.5), Methanol ( 3.4) und Wasser 94:4,5:1,5 (V/V/V). Trennkammer mit gesättigter Atmosphäre
3.18.4. Mischung aus Chloroform ( 3.2) und Methanol ( 3.4) 94:6 (V/V). Trennkammer mit gesättigter Atmosphäre.
3.18.5. Mischung aus Chloroform ( 3.2) und Methanol ( 3.4) 97:3 (V/V). Trennkammer mit gesättigter Atmosphäre
4. Geräte
4.1. Mahl- und Mischgerät
4.2. Schüttelmaschine oder Magnetrührer
4.3. Faltenfilter, Schleicher & Schüll Nr. 588 oder gleichwertige Filter, Durchmesser 24 cm
4.4. Chromatographierohr aus Glas, (innerer Durchmesser 22 mm, Länge 300 mm), mit Teflonhahn und 250-ml-Vorratsbehälter
4.5. Vacuumrotationsverdampfer mit 500-ml-Rundkolben
4.6. 500-ml-Erlenmeyerkolben mit Schliffstopfen
4.7. Ausrüstung für Dünnschicht-Chromatographie
4.8. Glasplatten für Dünnschicht-Chromatographie, 200 - 200 mm. Sie werden wie folgt vorbereitet (die angegebenen Mengen reichen für fünf Platten): 30 g Kieselgel G-HR ( 3.15) werden in einem Erlenmeyerkolben mit 60 ml destilliertem Wasser versetzt, der Kolben wird verschlossen und eine Minute geschüttelt. Die Suspension wird in einer einheitlichen Schichtdicke von 0,25 mm auf die Platten aufgetragen. Man lässt die Platten an der Luft trocknen und bewahrt sie dann im Exsikkator über Kieselgel als Trocknungsmittel auf. Vor Verwendung werden sie eine Stunde im Trockenschrank bei 110 °C aktiviert.
Fertigplatten können verwendet werden, sofern sie einen ähnlichen Trenneffekt ergeben wie die in der oben beschriebenen Weise hergestellten.
4.9. Analysenlampe für langwelligen UV-Bereich (360 nm). Die Lichtintensität muss ausreichen, um einen Aflatoxin-B1-Fleck von 1,0 ng auf einer Dünnschichtplatte in 10 cm Entfernung von der Lampe noch deutlich zu erkennen.
4.10. 10-ml-Reagenzgläser, graduiert, mit Schliff und Polyäthylenstopfen
4.11. UV-Spektralphotometer
4.12. Fluorodensitometer (wahlweise)
5. Ausführung
5.1. Vorbereitung der Probe (siehe unter Bemerkungen, Teil C, Punkt 1)
Die Probe wird gemahlen, so dass sie vollständig durch ein 1-mm-Sieb (gem. ISO-Empfehlung R 565) hindurchgeht.
5.2. Extraktion
50,0 g der gemahlenen und homogenisierten Probe werden in einen 500 ml-Erlenmeyerkolben ( 4.6) eingewogen, man fügt 25 g Kieselgur ( 3.14), 25 ml Wasser und 250 ml Chloroform ( 3.2) hinzu. Der Kolben wird verschlossen und 30 Minuten mit dem Gerät ( 4.2) geschüttelt bzw. gerührt. Anschließend filtriert man durch ein Faltenfilter ( 4.3), die ersten 10 ml des Filtrats werden verworfen, die nächsten 50 ml aufgefangen.
5.3. Säulenchromatographische Reinigung
Das untere Ende des Chromatographie-Rohrs ( 4.4) wird mit einem Watte- oder Glaswollepfropfen ( 3.9) versehen, das Rohr zu etwa zwei Dritteln mit Chloroform ( 3.2) gefüllt, dann fügt man 5 g Natriumsulfat ( 3.10) hinzu, wobei darauf zu achten ist, dass die Natriumsulfatschicht eine ebene Fläche bildet. Anschließend gibt man 10 g Kieselgel ( 3.8) in kleinen Portionen hinzu. Nach jeder Zugabe ist vorsichtig zu rühren, um Luftblasen zu entfernen. Man lässt das Kieselgel 15 Minuten absetzen und fügt dann vorsichtig 15 g Natriumsulfat ( 3.10) hinzu. Man lässt dann die Flüssigkeit bis unmittelbar an die Oberfläche des Natriumsulfats ablaufen.
50 ml des nach 5.2 erhaltenen Filtrats werden mit 100 ml n-Hexan ( 3.3) gemischt. Man gibt die Mischung quantitativ auf die. Säule und lässt die Flüssigkeit bis zur Oberfläche des Natriumsulfats einsickern. Anschließend fügt man 100 ml Diäthyläther ( 3.5) hinzu und lässt die Flüssigkeit wieder bis zur Oberfläche des Natriumsulfats einsickern. Die Durchflussgeschwindigkeit soll 8 bis 12 ml/ Minute betragen, ein Trockenlaufen der Säule ist zu vermeiden. Der Durchlauf wird verworfen. Man eluiert danach mit 150 ml Chloroform-Methanol-Gemisch ( 3.7), wobei das gesamte Eluat aufgefangen wird.
Das Eluat wird im Vakuumrotationsverdampfer ( 4.5) bei einer Temperatur von weniger als 50° C unter einem inerten Gasstrom ( 3.11) bis fast zur Trockne eingedampft. Der Rückstand wird mit Chloroform ( 3.2) oder Benzol-Acetonitril-Gemisch ( 3.6) quantitativ in ein 10-ml-Reagenzglas ( 4.10) überführt, die Lösung im inerten Gasstrom ( 3.11) eingeengt und anschließend mit Chloroform ( 3.2) oder Benzol-Acetonitril-Gemisch ( 3.6) auf ein Volumen von 2,0 ml aufgefüllt.
5.4. Dünnschicht-Chromatographie
Auf einer Dünnschicht-Platte ( 4.8) werden 2 cm vom unteren Rand entfernt nebeneinander im Abstand von 2 cm folgende Mengen der Standardlösung und des Extraktes punktförmig aufgetragen:
Die Platte wird vor Licht geschützt, mit einem der Fließmittel ( 3.18) entwickelt. Zur Auswahl eines geeigneten Fließmittels trägt man vorher 25 µl der qualitativen Standardlösung ( 3.17) auf eine der Platten auf und prüft, ob bei der Entwicklung eine vollständige Trennung der Aflatoxine B1 und B2 erreicht wird.
Man lässt das Fließmittel im Dunkeln verdampfen und betrachtet dann die Platte im UV-Licht in 10 cm Abstand von der Lampe ( 4.9). Die Flecke von Aflatoxin B1 weisen eine blaue Fluoreszenz auf.
5.5. Quantitative Bestimmung
Die Bestimmung ist entweder visuell oder durch Fluorodensitometrie, wie nachstehend beschrieben, vorzunehmen.
5.5.1. Visuelle Bestimmung
Die im Extrakt enthaltene Menge Aflatoxin B1 wird bestimmt, indem man die Fluoreszenzintensität der Flecke des Extrakts mit denen der Standardlösung vergleicht. Falls erforderlich, ist zu interpolieren. Die durch Übereinanderauftragen von Extrakt und Standardlösung erzielte Fluoreszenzintensität muss stärker sein als die von 10 µl des Extrakts und es darf nur ein einziger Fleck erkennbar sein. Ist die Fluoreszenz-Intensität von 10 µl Extrakt stärker als die von 40-µl-Standardlösung, so ist der Extrakt auf das 10- oder 100fache mit Chloroform ( 3.2) oder Benzol-Acetonitril-Gemisch ( 3.6) zu verdünnen und erneut in der beschriebenen Weise auf eine Dünnschichtplatte aufzutragen.
5.5.2. Durch Fluorodensitometrische Messung
Die Fluoreszenzintensität der Aflatoxin-B1-Flecke wird mit Hilfe des Fluorodensitometers ( 4.12) bei 443 nm gemessen bei einer Anregung mit 365 nm. Durch Vergleich der Fluoreszenzintensitäten von Extrakt- und Standardflecken bestimmt man die in den Extrakt-Flecken enthaltene Menge von Aflatoxin B1.
5.6. Identifizierung von Aflatoxin B1
Die Identifizierung von Aflatoxin B1im Extrakt erfolgt durch nachstehend beschriebene Verfahren.
5.6.1. Behandlung mit Schwefelsäure
Das nach 5.4 erhaltene Chromatogramm wird mit Schwefelsäure ( 3.13) besprüht. Die Fluoreszenz der Aflatoxin-B1-Flecke muss im UV-Licht von blau nach gelb umschlagen.
5.6.2. Zweidimensionale Chromatographie mit Bildung von Aflatoxin B1-hemiacetal (Aflatoxin B2a)
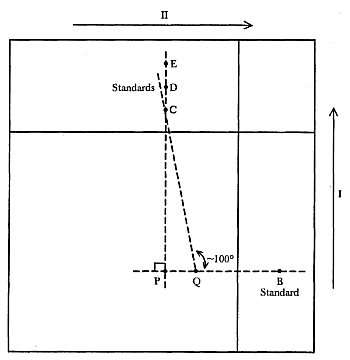
Anmerkung: Die nachfolgend beschriebenen Arbeitsgänge müssen genau nach der in Abbildung 3 wiedergegebenen Skizze durchgeführt werden.
5.6.2.1. Auftragen der Lösungen
In eine Platte ( 4.8) werden, parallel zu zwei angrenzenden Seiten (jeweils 6 cm vom Rand entfernt), zwei gerade Linien als Begrenzung für die Lösungsmittelfronten geritzt. Auf die Platte trägt man mit Hilfe einer Kapillarpipette oder einer Mikroliterspritze folgende Lösungen auf
5.6.2.2. Entwicklung
Das Chromatogramm wird mit dem Fließmittel ( 3.18.1) (1-cm-Schicht in einer Trennkammer mit nicht gesättigter Atmosphäre) im Dunkeln in Richtung I entwickelt bis die Lösungsmittelfront die Begrenzungslinie erreicht. Man nimmt danach die Platte aus der Trennkammer und lässt sie im Dunkeln fünf Minuten bei Raumtemperatur trocknen. Während man den Rest der Platte mit einer Glasscheibe abdeckt, besprüht man einen 2,5 cm breiten Streifen (schraffierte Fläche in der Abbildung 3), der die Punkte a und B überdeckt, mit Salzsäure ( 3.12), bis eine Dunkelfärbung auftritt. Man lässt 10 Minuten im Dunkeln reagieren und trocknet die Platte dann in einem Luftstrom bei Raumtemperatur.
Anschließend entwickelt man das Chromatogramm mit dem Fließmittel ( 3.18.1) (1-cm-Schicht in einer Trennkammer mit ungesättigter Atmosphäre) in Richtung II, bis die Lösungsmittelfront die Begrenzungslinie erreicht. Man nimmt die Platte aus der Trennkammer und lässt sie bei Raumtemperatur trocknen.
5.6.2.3. Interpretation des Chromatogramms
Man betrachtet das Chromatogramm unter UV-Licht ( 4.9) und prüft auf nachfolgende Merkmale:
6. Berechnung der Ergebnisse
6.1. Visuelle Messung
Der Aflatoxin-B1-Gehalt der Probe in Mikrogramm pro kg (ppb) wird nach folgender Formel berechnet:
(S · Y · V) / (W · X)
Hierbei sind:
Y und X die Mengen - in Mikroliter - der Aflatoxin-B1-Standardlösung ( 3.16) bzw. des Extrakts, deren Fluoreszenz-Intensität identisch ist.
S = Konzentration in Mikrogramm Aflatoxin B1 je ml der Standardlösung ( 3.16);
V = Endvolumen des Extrakts in Mikroliter, unter Berücksichtigung etwaiger Verdünnung;
W = Gewicht der Probe in Gramm, bezogen auf die für die säulenchromatographische Reinigung verwendete Extraktmenge.
6.2. Messungen durch Fluorodensitometrie
Der Gehalt an Aflatoxin B1 der Probe in Mikrogramm pro kg (ppb) wird nach folgender Formel berechnet:
(S · V) / (W · Y)
Hierbei sind:
Y = Volumen des auf die Platte aufgetragenen Extrakts in Mikroliter (10 oder 20 µl);
S = Aflatoxin-B1-Gehalt des Extraktflecks in Nanogramm, den man aus der Messung erhält (bezogen auf das angewendete Volumen Y);
V = Endvolumen des Extrakts in Mikroliter, unter Berücksichtigung etwaiger Verdünnung;
W = Gewicht der Probe in Gramm, bezogen auf die für die säulenchromatographische Reinigung verwendete Extraktmenge.
7. Herstellung und Kontrolle der Standardlösung ( 3.16)
7.1. Bestimmung der Aflatoxin-B1-Konzentration
Eine Aflatoxin-B1-Standardlösung wird in Chloroform ( 3.2) oder Benzol-Acetonitril-Gemisch ( 3.6) mit einer Konzentration von 8 bis 10 µg je ml hergestellt. Das Absorptionsspektrum wird zwischen 330 und 370 nm mit einem Spektralphotometer ( 4.11) gemessen.
Die Extinktion (A) ist im Fall einer Chloroformlösung bei 363 nm, im Fall einer Lösung in Benzol-Acetonitril-Gemisch bei 348 nm zu messen.
Die Aflatoxin-B1-Konzentration in Mikrogramm je ml Lösung wird nach folgenden Formeln berechnet:
(312 · a · 1000) / 20.600
bei der Chloroformlösung;
(312 · a · 1000) / 19.800
bei der Lösung im Benzol-Acetonitril-Gemisch.
Die zur Herstellung einer Standardarbeitslösung mit einer Aflatoxin-B1-Konzentration von etwa 0,1 µg je ml erforderlichen Verdünnungen werden im Dunkeln vorgenommen. Diese Lösung ist, bei 4° C im Kühlschrank aufbewahrt, zwei Wochen haltbar.
7.2. Kontrolle der chromatographischen Reinheit
Auf einer Dünnschichtplatte ( 4.8) werden 5 µl der Aflatoxin-B1-Standardlösung, die8-10 0 µg g Aflatoxin B1je ml enthält (siehe 7.1) aufgetragen. Das Chromatogramm wird nach 5.4 entwickelt. Im UV-Licht darf nur ein einziger Fleck erkennbar sein; außerdem darf an der Stelle der ursprünglichen Auftragung keine Fluoreszenz wahrnehmbar sein.
8. Wiederholbarkeit
Der Unterschied zwischen den Ergebnissen zweier Parallelbestimmungen eines Analytikers sollte in derselben Probe bei Gehalten von
9. Vergleichbarkeit
Siehe unter Bemerkungen, Teil C, Punkt 2.
B. Bestimmung von Aflatoxin B1 durch Hochleistungsflüssigkeitschromatographie
1. Zweck und Anwendungsbereich
Die Methode dient der Bestimmung von Aflatoxin B1 in Futtermitteln, einschließlich solchen, die Citrustrester enthalten. Die untere Grenze der Bestimmbarkeit beträgt 0,001 mg/kg (1 ppb).
2. Prinzip
Die Probe wird mit Chloroform extrahiert. Der Extrakt wird filtriert und ein allquoter Teil über eine Florisil-Kartusche und anschließend über eine C18-Kartusche gereinigt. Die endgültige Trennung und Bestimmung erfolgen mit einer C18-Umkehrphasen-HPLCSäule und anschließender Nachsäulenderivatisierung mit Jod in Wasser sowie Fluoreszenzdetektion.
Anmerkung: Mykotoxine sind äußerst toxische Substanzen. Alle Arbeiten sollten in einem dafür vorgesehenen Abzug durchgeführt werden. Besondere Vorsichtsmaßnahmen sollten getroffen werden, wenn die Toxine in fester Form vorliegen, wegen ihrer elektrostatischen Eigenschaft und der daraus resultierenden Tendenz, sich auf Arbeitsflächen zu verteilen.
3. Reagenzien
3.1. Chloroform, stabilisiert mit 0,5 bis 1,0 % Ethanol (m/m). Siehe Bemerkung 10.2.
3.2. Methanol, für die HPLC zur Herstellung von 3.6.
3.3. Aceton.
3.4. Acetonitril für die HPLC.
3.5. Elutionsmittel: Sie werden einen Tag vor der Verwendung bereitet, oder die Luft in den Lösemitteln wird mit Hilfe von Ultraschall entfernt.
3.5.1. Mischung aus Aceton ( 3.3) und Wasser, 98 + 2 (V + V).
3.5.2. Mischung aus Wasser und Methanol ( 3.2), 80 + 20 (V + V).
3.5.3. Mischung aus Wasser und Aceton (3.3), 85 + 15 (V + V).
3.6. Mobile Phase für die HPLC.
Mischung aus Wasser, Methanol ( 3.2) und Acetonitril ( 3.4), 130 + 70 + 40 (V + V + V).
N.B.: Gegebenenfalls muss das Mischungsverhältnis der mobilen Phase der Charakteristik der verwendeten HPLC-Säule angepaßt werden.
3.7. Gesättigte Jodlösung: 2 g Jod werden in 400 ml Wasser gegeben, mindestens 90 Minuten gemischt und durch ein Membranfilter ( 4.15) filtriert. Die gesättigte Lösung ist vor Licht zu schützen, um einen fotochemischen Abbau zu verhindern.
3.8. Celite 545, mit Säure gewaschen, oder gleichwertiges Material.
3.9. Florisil-Kartusche (Waters SEP-PAK) oder gleichwertiges Material.
3.10. C18-Kartusche (Waters SEP-PAK) oder gleichwertiges Material.
3.11. Inertgas, z.B. Stickstoff.
3.12. Aflatoxin-B1-Standardlösung in Chloroform, Konzentration 10 µg/ ml.
Die Konzentration der Lösung wird folgendermaßen überprüft: Bestimmung des Absorptionsspektrums der Lösung zwischen 330 und 370 nm mit Hilfe des Spektralphotometers ( 4.23). Messung der Extinktion (E) bei 363 nm. Berechnung der Konzentration von Aflatoxin B1 in 1 µg/ml mit der Formel:
Konzentration (èmg=ml ) = (312 x E x 1.000) / 22.300 = 13; 991 x E
3.12.1. Aflatoxin-B1-Stammlösung in Chloroform.
2,5 ml der Aflatoxin-B1-Standardlösung ( 3.12) werden in einen 50-ml-Messkolben überführt und der Kolben mit Chloroform ( 3.1) zur Marke aufgefüllt. Diese Stammlösung wird kühl (4 °C), dunkel, dicht verschlossen und mit Alufolie umwickelt aufbewahrt.
3.13. Aflatoxin-B1-Eichlösungen für die HPLC.
Anmerkung: Für die Herstellung dieser Lösungen sind säuregewaschene Glasgeräte zu verwenden (siehe 4, Geräte).
3.13.1. Eichlösung, 4 ng/ml.
Man lässt den mit Alufolie umwickelten Kolben mit der Stammlösung ( 3.12.1) auf Raumtemperatur erwärmen (einige Stunden). 400 µl der Stammlösung (200 ng Aflatoxin B1) werden in einen 50-ml-Messkolben überführt und in einem Strom von Inertgas ( 3.11) zur Trockne eingedampft. Der erhaltene Rückstand wird in ca. 20 ml Wasser-Aceton-Gemisch ( 3.5.3) gelöst, bis zur Marke mit Wasser-Aceton-Gemisch ( 3.5.3) aufgefüllt und gut durchmischt.
3.13.2. Eichlösung, 3 ng/ml.
7,5 ml der Eichlösung ( 3.13.1) werden quantitativ in einen 10-ml-Messkolben überführt. Es wird mit Wasser-Aceton-Gemisch ( 3.5.3) zur Marke aufgefüllt und der Kolbeninhalt gut durchmischt.
3.13.3. Eichlösung, 2 ng/ml.
25 ml der Eichlösung ( 3.13.1) werden quantitativ in einen 50-ml-Messkolben überführt. Es wird mit Wasser-Aceton-Gemisch ( 3.5.3) zur Marke aufgefüllt, und der Kolbeninhalt wird gut durchmischt.
Diese Lösung gilt auch als Bezugsstandard, der für die wiederholte Einspritzung während der HPLC-Bestimmung ( 5.5) verwendet wird.
3.13.4. Eichlösung, 1 ng/ml.
2,5 ml Eichlösung ( 3.13.1) werden quantitativ in einen 10-ml-Messkolben überführt. Es wird mit Wasser-Aceton-Gemisch ( 3.5.3) zur Marke aufgefüllt und der Kolbeninhalt gut durchmischt.
3.14. Ampulle mit einer Mischung der Aflatoxine B1, B2, G1 und G2 in Konzentrationen von ca. 1; 0,5; 1 bzw. 0,5 µg/ml in 1 ml Chloroform.
3.14.1. Testlösung für die HPLC.
Der Inhalt der Ampulle ( 3.14) wird in ein Reagenzglas mit Schliffstopfen oder Schraubverschluss überführt. 40 µl dieser Lösung werden in einen 10-ml-Messkolben oder in ein Reagenzglas mit Schliffstopfen (mit Säure gewaschen) ( 4.22) überführt. Nach Abdampfen des Chloroforms im Inertgasstrom ( 3.11) wird der Rückstand in 10 ml Wasser-Aceton-Gemisch ( 3.5.3) gelöst.
3.15. Reagenzien für die Identitätsbestätigung ( 6).
3.15.1. Gesättigte Natriumchloridlösung.
3.15.2. Natriumsulfat, wasserfrei, gekörnt.
4. Geräte
Achtung: Die Verwendung von Glasgeräten, die nicht mit Säure gewaschen wurden, kann bei wässrigen Aflatoxinlösungen zu Verlusten führen. Besondere Sorgfalt ist bei neuen Glasgeräten und Einweg-Glasgeräten wie Ampullen für automatische Probengeber und Pasteurpipetten geboten. Daher müssen Laborgeräte aus Glas, die mit wässrigen Aflatoxinlösungen in Berührung kommen, mehrere Stunden in verdünnter Säure (z.B. Schwefelsäure, c = 2 mol/l) liegen und dann zur Beseitigung sämtlicher Säurereste sorgfältig mit destilliertem Wasser gespült werden (z.B. dreimal, mit pH-Papier prüfen). In der Praxis ist diese Behandlung erforderlich bei Rundkolben ( 4.4), Messkolben, Messzylindern, Reagenzgläsern oder Gefäßen, die für Eichlösungen und Endextrakte verwendet werden (insbesondere bei Ampullen für automatische Probengeber) sowie für Pasteurpipetten, die zur Überführung von Eichlösungen und Extrakten benutzt werden.
4.1. Mahl- und Mischgerät.
4.2. Sieb mit einer Maschenweite von 1,0 mm (ISO R 565).
4.3. Schüttelmaschine.
4.4. Vakuumrotationsverdampfer mit 150- bis 250-ml-Rundkolben.
4.5. Hochleistungs-Flüssigkeitschromatograph, Injektor mit Probenschleife, geeignet für die Injektion von 250 µl. Die Angaben des Herstellers für teilweise oder vollständige Schleifenfüllung sind zu beachten.
4.6. HPLC-Trennsäule: C18-Füllmaterial, Teilchengröße 3 µm oder 5 µm.
4.7. Pulsationsfreie Pumpe für das Nachsäulen-Jodreagenz.
4.8. Totvolumenfreies T-Stück (Valco) aus Edelstahl, (1/16" × 0,75 mm).
4.9. Spiralreaktionsrohr aus Teflon oder Edelstahl. Abmessungen von 3.000 × 0,5 mm bis 5.000 × 0,5 mm sind in Verbindung mit 5 µm oder 3 µm HPLC-Säulen erwiesenermaßen geeignet.
4.10. Thermostatgesteuertes, auf 60 °C eingestelltes Wasserbad, Temperaturregelung genauer als 0,1 °C.
4.11. Fluoreszenzdetektor mit 365 nm Anregungs- und 435 nm Emissionswellenlänge (bei Filtergeräten: Emissionswellenlänge > 400 nm).
Ein Nachweis von mindestens 0,05 ng Aflatoxin B1 solltemöglich sein. Unter Umständen kann ein Gegendruck erforderlich sein (z.B. Restriktor bzw. Teflon- oder Edelstahlrohr ausreichender Länge am Detektorauslass), um Luftblasen in der Durchflusszelle zu vermeiden.
4.12. Schreiber.
4.13. Elektronischer Integrator (wahlweise).
4.14. Faltenfilter, Durchmesser 24 cm, Macherey-Nagel 617 1/4, oder gleichwertiges Filter.
4.15. Membran-Filter mit einer Porengröße von 0,45 µm, Millipore (H.A.W.P. 04700) oder gleichwertiges Filter.
4.16. 500-ml-Erlenmeyerkolben mit Schliffstopfen.
4.17. Glassäule, Innendurchmesser ca. 1 cm, Länge ca. 30 cm, mit Luer-Endstück.
4.18. Chloroformbeständiger Luer-Absperrhahn aus Nylon (z.B. BIORAD 7328017; Analytichem A1 6078; J. T. Baker 4514).
4.19. Chemisch inerte Spritze, 10 ml, mit Luer-Anschluss.
4.20. Spritze geeignet für Injektionen von 250 µl (siehe 4.5).
4.21. 100-µl-Spritze für die Herstellung der Eichlösungen. Durch Wägung ist zu überprüfen, dass die Genauigkeit innerhalb von 2 % liegt.
4.22. 10-ml-Reagenzgläser, graduiert, mit Schliffstopfen.
4.23. UV-Spektralphotometer.
4.24. Geräte für die Identitätsbestätigung ( 6).
4.24.1. Mit Säure gespülter 100-ml-Scheidetrichter mit Teflon-Absperrhahn.
4.24.2. Heizblock, 40-50 °C.
5. Ausführung
5.1. Vorbereitung der Probe
Die Probe wird gemahlen, so dass sie vollständig durch das Sieb ( 4.2) hindurchgeht.
5.2. Untersuchungsprobe
50 g der vorbereiteten Probe werden in einen Erlenmeyerkolben ( 4.16) eingewogen.
5.3. Extraktion
Zu der Untersuchungsprobe ( 5.2) werden 25 g Celite ( 3.8), 250 ml Chloroform ( 3.1) und 25 ml Wasser gegeben. Der Kolben wird verschlossen, und es wird 30 Minuten mit einer Schüttelmaschine ( 4.3) gemischt. Anschließend wird durch ein Faltenfilter ( 4.14) filtriert, und 50 ml des Filtrats werden aufgefangen. Erforderlichenfalls wird ein aliquoter Teil des Filtrats mit Chloroform auf 50 ml verdünnt, so dass die Konzentration von Aflatoxin B1 nicht größer als 4 ng/ml ist.
5.4. Reinigung
Die Reinigung sollte ohne nennenswerte Unterbrechungen durchgeführt werden.
Achtung:
Aflatoxinhaltige Lösungen müssen so gut wie möglich vor Licht geschützt werden (Aufbewahrung im Dunkeln, Verwendung von Alufolie).
5.4.1. Florisil-SEP-PAK-Reinigung
5.4.1.1. Vorbereitung der Säulen-Kartuschen-Kombination.
Am kürzeren Ende der Florisil-Kartusche ( 3.9) wird ein Absperrhahn ( 4.18) angebracht (siehe Abbildung 1). Die Kartusche wird gewaschen und die Luft entfernt, indem von 10 ml Chloroform ( 3.1) 8 ml mit einer Spritze ( 4.19) über den Absperrhahn schnell durch die Kartusche hineingedrückt werden. Das längere Ende der Kartusche wird mit einer Glassäule ( 4.17) verbunden und die verbleibenden 2 ml Chloroform durch die Kartusche in die Glassäule gedrückt. Der Absperrhahn wird geschlossen und die Spritze entfernt.
5.4.1.2. Reinigung.
Das unter 5.3 gewonnene Filtrat wird in die Säulen-Kartuschen-Kombination gegeben, und man lässt es durch Schwerkraft ablaufen. Es wird mit 5 ml Chloroform ( 3.1) und anschließend mit 20 ml Methanol ( 3.2) gespült. Die Eluate werfen verworfen. Dabei ist ein Trockenlaufen der Säulen-Kartuschen-Kombination zu vermeiden. Das Aflatoxin B1 wird mit 40 ml Aceton-Wasser-Gemisch ( 3.5.1) eluiert und das gesamte Eluat im Rundkolben des Vakuum-Rotationsverdampfers ( 4.4) aufgefangen. Das Eluat wird am Vakuum-Rotationsverdampfer bei 40-50 °C eingeengt, bis kein Aceton mehr übergeht. (Anmerkung: etwa 0,5 ml der Flüssigkeit verbleiben dabei im Kolben. Versuche haben ergeben, dass ein weiteres Abdampfen sich nicht nachteilig auswirkt und dass die verbleibenden 0,5 ml Flüssigkeit keine nennenwerte Acetonmenge enthalten. Acetonreste können zu Verlusten von Aflatoxin B1 in der C18-Kartuscheführen.)
Nach Zugabe von 1 ml Methanol ( 3.2) wird der Kolben geschwenkt, um das Aflatoxin B1 an den Gefäßwänden zu lösen. Anschließend werden 4 ml Wasser zugegeben, und der Kolbeninhalt wird durchmischt. Die Kartusche wird abgenommen und verworfen. Die Glassäule wird mit Wasser gespült und für den Reinigungsschnitt mit C18 bereitgehalten.
5.4.2. C18-SEP-PAK-Reinigung
5.4.2.1. Vorbereitung der Säule-Kartuschen-Kombination.
Das kürzere Ende einer C18-Kartusche ( 3.10) wird mit einem Absperrhahn (4.18) versehen (siehe Abbildung 1). Die Kartusche wird vorbereitet und die Luft entfernt, indem mit einer Spritze ( 4.19) 10 ml Methanol ( 3.2) schnell über den Absperrhahn durch die Kartusche gedrückt werden. (Luftblasen in der Kartusche sind als helle Flecken vor dem ansonsten gräulichen Hintergrund erkennbar). Von 10 ml Wasser werden 8 ml durch die Kartusche gedrückt. (Beim Übergang von Methanol auf Wasser ist das Eindringen von Luft in die Kartusche zu vermeiden.) Das längere Ende der Kartusche wird mit einer Glassäule ( 4.17) verbunden und die restlichen 2 ml durch die Kartusche in die Glassäule gedrückt. Der Absperrhahn wird geschlossen und die Spritze entfernt.
5.4.2.2. Reinigung.
Der unter 5.4.1.2 erhaltene Exrakt wird quantitativ in die Glassäule ( 4.17) überführt. Der Kolben wird zweimal mit 5 ml Wasser-Methanol-Gemisch ( 3.5.2) gespült, und es wird durch Schwerkraft ablaufen gelassen. Während dieses Arbeitsgangs ist ein Trockenlaufen der Säulen-Kartuschen-Kombination zu vermeiden. (Wenn sich in der Verengung nahe der Kartusche Luftblasen bilden, wird der Fluss unterbrochen, und die Luftblasen werden durch leichtes Klopfen an das obere Ende der Glassäule entfernt.) Anschließend wird mit 25 ml Wasser-Methanol-Gemisch ( 3.5.2) eluiert, und das Eluat wird verworfen. Das Aflatoxin B1 wird mit 50 ml Wasser-Aceton-Gemisch ( 3.5.3) eluiert und das gesamte Eluat in einem 50-ml-Messkolben aufgefangen. Der Kolben wird mit Wasser bis zur Marke aufgefüllt und der Kolbeninhalt gut durchmischt. Die so erhaltene Lösung wird für die HPLC ( 5.5) verwendet.
Achtung: Eine Filtration des Endextrakts vor der HPLC ist normalerweise nicht erforderlich. Wird dies für notwendig gehalten, dürfen keine Cellulosefilter verwendet werden, da diese zu Aflatoxin-B1-Verlusten führen können. Teflonfilter können verwendet werden.
5.5. Hochleistungsflüssigkeitschromatographie
(Aufbau der Anlage siehe Abbildung 2.) Es ist genügend Zeit zur Konditionierung und Stabilisierung der Geräte vorzusehen.
Anmerkung 1:
Die Flussraten für die mobile Phase und das Nachsäulenreagenz sind nur Anhaltspunkte. Sie sind der Art und Dimension der HPLC-Säule entsprechend anzupassen.
Anmerkung 2:
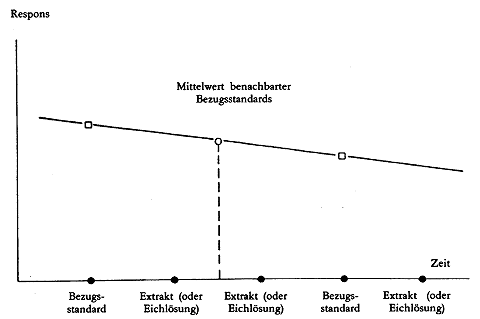
Der Detektorresponse des Aflatoxins B1 ist temperaturabhängig, daher sollten Korrekturen für die Abweichungen vorgenommen werden (siehe Abbildung 3). Durch Einspritzung einer bestimmten Menge des Aflatoxin-B1-Bezugsstandards ( 3.13.3) in regelmäßigen Abständen (z.B. jede dritte Einspritzung) können die Aflatoxin-B1-Peakwerte zwischen diesen Bezugsstandards mit Hilfe des mittleren Responses korrigiert werden. Voraussetzung ist, dass der Unterschied der Response aufeinanderfolgenden Bezugsstandards sehr klein (10 %) ist. Daher müssen die Einspritzungen ohne Unterbrechung erfolgen. Ist eine Unterbrechung notwendig, muss die letzte Einspritzung vor der Unterbrechung und die erste danach mit dem Bezugsstandard ( 3.13.3) durchgeführt werden. Da die Eichkurve linear ist und durch den Nullpunkt verläuft, werden die Aflatoxin-B1-Mengen in den Probenextrakten unmittelbar mit Hilfe der benachbarten Standards bestimmt.
5.5.1. Einstellung der HPLC-Pumpe
Die HPLC-Pumpe ( 4.5) wird so eingestellt, dass sich bei Verwendung der mobilen Phase ( 3.6) eine Flussrate von 0,5 bzw. 0,3 ml/ min für die5-µm- bzw. 3-µm-HPLC-Säule ( 4.6) ergibt.
5.5.2. Einstellung der Nachsäulenpumpe
Die Pumpe ( 4.7) wird so eingestellt, dass sich eine Flussrate von 0,2 bis 0,4 ml/min der gesättigten Jod-Wasser-Lösung ( 3.7) ergibt. Als grober Anhaltspunkt können Flussraten von ca. 0,4 bzw. 0,2 ml/min bei Flussraten von 0,5 bzw. 0,3 ml/min der mobilen Phase ( 3.6) empfohlen werden.
5.5.3. Fluoreszenzdetektor
Der Fluoreszenzdetektor ( 4.11) wird auf eine Anregungswellenlänge von 365 nm und eine Emissionswellenlänge von 435 nm (Filterinstrument: > 400 nm) eingestellt. Die Detektorabschwächung wird so eingestellt, dass 1 ng Aflatoxin B1 ca 80 % Vollausschlag des Schreibers ergibt.
5.5.4. Injektor
Von allen Lösungen werden Volumina von 250 µl gemäß den Herstellerangaben eingespritzt.
5.5.5. Überprüfung der chromatographischen Trennung
Die Testlösung für die Chromatographie ( 3.14.1) wird eingespritzt. Der Abstand der Täler sollte weniger als 5 % der Summe der Peakhöhlen der benachbarten Peaks betragen.
5.5.6. Überprüfung der Systemstabilität
Vor jeder Analysenreihe wird der Bezugsstandard ( 3.13.3) wiederholt eingespritzt, bis konstante Peakflächen erhalten werden.
Anmerkung:
Die Peakflächen von Aflatoxin B1 sollten bei aufeinanderfolgenden Einspritzungen um nicht mehr als 6 % voneinander abweichen. Es ist unverzüglich mit der Überprüfung der Linearität ( 5.5.7) fortzufahren.
5.5.7. Überprüfung der Linearität
Die Aflatoxin-B1-Eichlösungen ( 3.13.1 bis 3.13.4) werden eingespritzt. Für jede dritte Einspritzung ist der Bezugsstandard 3.13.3 zu verwenden, um Responseabweichungen zu korrigieren (Anmerkung: Die Peak-Response für diesen Bezugsstandard dürfen nicht mehr als 10 % innerhalb von 90 Minuten voneinander abweichen.) Die Abweichungen werden gemäß der Formel in Punkt 7 korrigiert. Die Eichkurve sollte linear sein und innerhalb des zweifachen Standardfehlers des y-Wertes durch den Nullpunkt verlaufen. Die ermittelten Werte dürfen nicht mehr als 3 % von den Nennwerten abweichen. Sind diese Anforderungen erfüllt, ist unverzüglich mit Punkt 5.5.8 fortzufahren; wenn nicht, sind vorher die Fehlerquellen festzustellen und zu beseitigen.
5.5.8. Einspritzung der Probenextrakte
Die gereinigten Probenextrakte ( 5.4.2.2) werden eingespritzt. Nach je zwei Probenextrakten ist die Einspritzung des Bezugsstandards ( 3.13.3) zu wiederholen, also: Bezugsstandard, Extrakt Extrakt, Bezugsstandard, Ektrakt, Ektrakt, Bezugsstandard usw.
6. Identitätsbestätigung
6.1. Weitere Behandlung des Extraktes (5.4.2.2)
Dem nach 5.4.2.2 gewonnen Endextrakt werden 5 ml Natriumchloridlösung ( 3.15.1) zugesetzt. Man schüttelt in einem Scheidetricher ( 4.24.1) dreimal mit je 2 ml Chloroform ( 3.1) jeweils eine Minute. Die vereinigten Chloroformextrakte werden über ca. 1 g Natriumsulfat ( 3.15.2) in ein 10-ml-Reagenzglas gegossen. Es kann ein kleiner Trichter (Durchmesser 4 cm) verwendet werden, in dessen Auslauf sich ein Wattepfropf, der mit 1 g Natriumsulfat ( 3.15.2) bedeckt ist, befindet.
Die Natriumsulfatschicht wird mit einigen ml Chloroform ausgewaschen und die Waschlösung im gleichen Reagenzglas aufgefangen. Der Chloroformextrakt wird in diesem Reagenzglas in einem Heizblock ( 4.24.2) zur Trockne eingedampft und erneut in 1 ml Chloroform gelöst.
6.2. Herstellung der Derivate und Dünnschichtchromatographie
Vergleiche Richtlinie 76/372/EWG des Rates, Anhang, Methode a unter Punkt 5.6.2.
7. Berechnung der Ergebnisse
Der Aflatoxin-B1-Gehalt der Probe (µg/kg) wird mit folgender Formel berechnet:
Aflatoxin-B1-Gehalt in èmg/kg =
Hierbei sind:
m = die Mengean Aflatoxin B1 in ng, dargestellt durch den B1-Peak der Probe, die folgendermaßen berechnet wird:

P(Probe) = Peakfläche des Aflatoxins B1 der Probe
P(st1) = Peakfläche des Aflatoxins B1 bei der vorangegangenen Einspritzung des Bezugsstandards ( 3.13.3)
P(st2) = Peakfläche des Aflatoxins B1 bei der nachfolgenden Einspritzung des Bezugsstandards ( 3.13.3)
r(st) = eingespritzte Menge Aflatoxin B1 im Bezugsstandard ( 3.13.3) in ng
Vm = Volumen des eingespritzten Probenextrakts in ml
Ve x t = Endvolumen des Probenextrakts in ml, unter Berücksichtigung der Verdünnung ( 5.3)
M = Probeneinwaage in g
Vf= Volumen des auf die Florisilkartusche ( 5.4.1.2) gegebenen Filtrats in ml
Vc = Volumen des zur Extraktion der Probe verwendeten Chloroforms in ml
Wird dem in diesem Protokoll angegebenen Verfahren gefolgt, kann die Formel folgendermaßen verkürzt werden:
Aflatoxin-B1-Gehalt in µg/kg = 20 × m
7.1. Die Ergebnisse können auch durch Messung der Peak-Höhen ermittelt werden.
8. Wiederholbarkeit
Siehe 10.1 (Bemerkungen).
9. Vergleichbarkeit
Siehe 10.1 (Bemerkungen).
10. Bemerkungen
10.1. Reproduzierbarkeit
Eine auf internationaler Ebene durchgeführte Ringuntersuchung 9 an Mischfuttermitteln ergab hinsichtlich der Wiederholbarkeit und Vergleichbarkeit die in Tabelle 1 aufgeführten Ergebnisse. Der hier verwendete Begriff Wiederholbarkeit (r) bezeichnet das größte Verhältnis, das bei 95%iger Wahrscheinlichkeit im Vergleich von 2 Ergebnissen der gleichen Probe und unter gleichen Laborbedingungen nicht signifikant ist. Der Begriff Vergleichbarkeit (R) ist ähnlich definiert für den Vergleich zweier verschiedener Laboratorien. Gemäß ISO 3534 - 1977, Ziffer 2.35 10 und der Entscheidung 89/610/EWG der Kommission 11 sind in Tabelle 1 r und R auch als Variationskoeffizienten angegeben.
Tabelle 1 Wiederholbarkeit (r) und Vergleichbarkeit (R), ausgedrückt als Verhältniszahlen und als Variationskoeffizienten
(15 Laboratorien)
| Niveau | r | R | CVr * | CVR * |
| (µg/kg) | (%) | (%) | ||
| 8 und 14 | 1,4 | 1,7 | 11 | 18 |
| *) CV = Variationskoeffizient. | ||||
10.2. Stabilisierung des Chloroforms ( 3.1)
Die Absorptionseigenschaften der Florisilkartusche (3.9) können sich verändern, wenn ein anderer Stabilisator als Ethanol verwendet wird. Dieses sollte gemäß Punkt 10.3 überprüft werden für den Fall, dass das beschriebene Chloroform nicht erhältlich ist.
10.3. Richtigkeit
Die korrekte Durchführung des Verfahrens ist zu überprüfen, indem wiederholte Bestimmungen an zertifizierten Referenzmaterialien durchgeführt werden. Sind diese nicht verfügbar, sollte die Leistungsfähigkeit des Verfahrens durch Wiederfindungsversuche an dotierten Blindproben überprüft werden. Die Abweichung des Mittelwerts vom Sollwert, ausgedrückt in Prozenten des Sollwerts, soll einen Rahmen von -20 bis + 10 % nicht überschreiten.
Abbildung 1 Säulen-Kartuschen-Kombination
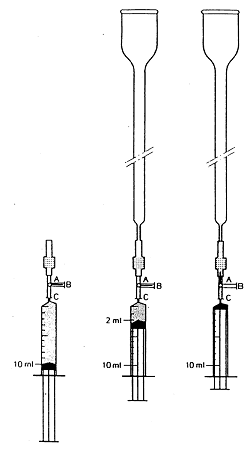
Abbildung 2: Flussschema des LC-Systems mit Einrichtung zur Nachsäulenderivatisierung mit Jod

Abbildung 3: Korrektur der Abweichung des Aflatoxin-B1-Responses durch Einspritzung des Bezugsstandards (3.13.3) in regelmäßigen Abständen
C. Bemerkungen betreffend Methode a und B
1. Entfettung
Proben mit mehr als 5 v. H. Fett sind nach der unter 5.1 beschriebenen Vorbereitung mit Petrolether (Kp 40 - 60 °C) zu entfetten.
In diesem Fall sind die Ergebnisse auf das Gewicht der nicht entfetteten Originalprobe zu beziehen.
2. Vergleichbarkeit der Ergebnisse der Methode A
Die Vergleichbarkeit der Ergebnisse, das heißt die Abweichung zwischen den von zwei oder mehr Laboratorien für die gleiche Probe erhaltenen Ergebnissen, wurde wie folgt bewertet:
± 50 v. H. des Mittelwertes der Ergebnisse bei Mittelwerten an Aflatoxin B1 von 10 bis 20 µg/kg;
± 10 µg/kg vom Mittelwert bei Mittelwerten von mehr als 20 bis 50 µg/kg;
± 20 v. H. des Mittelwertes bei Mittelwerten von mehr als 50 µg/kg.
| Anlage |
:____________________
1) ABl. Nr. L 170 vom 03.08.1970 S. 2.
2) ABl. Nr. L 73 vom 27.03.1972 S. 14.
3) ABl. Nr. L 155 vom 12.07.1971 S. 13.
4) ABl. Nr. L 279 vom 20.12.1971 S. 7.
5) ABl. Nr. L 123 vom 29.05.1972 S. 6.
6) ABl. Nr. L 83 vom 30.03.1973 S. 21.
7) ABl. Nr. L 108 vom 22.04.1974 S. 7.
8) ABl. Nr. L 32 vom 05.02.1975 S. 26.
9) Egmond, H.P. van, Heisterkamp, S.H. und Paulsch, W.E. (1991). Food Additives and Contaminants 8, S. 17-29.
10) ISO 3534-1977.
11) ABl. Nr. L 351 vom 02.12.1989 S. 39.
 |
ENDE | |
(Stand: 21.03.2024)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: ab 105.- € netto
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion