 Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 1970 -75, Lebensmittel - Futtermittel

| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 1970 -75, Lebensmittel - Futtermittel |
|
Dritte Richtlinie 72/199/EWG der Kommission vom 27. April 1972 zur Festlegung gemeinschaftlicher Analysemethoden für die amtliche Untersuchung von Futtermitteln
(ABl. Nr. L 123 vom 29.05.1972 S. 6, ber. 1980 L 320 S. 43;
RL 81/680/EWG - ABl. L 246 vom 30.07.1981 S. 32;
RL 84/4/EWG - ABl. Nr. L 15 vom 18.01.1984 S. 28;
RL 93/28/EWG - ABl. Nr. L 179 vom 22.07.1993 S. 8;
RL 98/54/EG - ABl. Nr. L 208 vom 24.07.1998 S. 49;
RL 1999/79/EG - ABl. Nr. L 209 vom 07.08.1999 S. 23;
VO (EG) 152/2009 - ABl. Nr. L 54 vom 26.02.2009 S. 1aufgehoben)
aufgehoben/ersetztgem. Art. 6 der VO (EG) 152/2009- Entsprechungstabelle
Die Kommission der Europäischen Gemeinschaften -
gestützt auf den Vertrag zur Gründung der Europäischen Wirtschaftsgemeinschaft,
gestützt auf die Richtlinie des Rates vom 20. Juli 1970 über die Einführung gemeinschaftlicher Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln 1, insbesondere auf Artikel 2,
in Erwägung nachstehender Gründe:
Die oben genannte Richtlinie bestimmt, dass die amtlichen Untersuchungen von Futtermitteln zur Feststellung, ob die auf Grund der Rechts- oder Verwaltungsvorschriften festgelegten Anforderungen hinsichtlich Beschaffenheit und Zusammensetzung der Futtermittel erfüllt sind, nach gemeinschaftlichen Probenahmeverfahren und Analysemethodendurchgeführt werden.
Die Richtlinien Nr. 71/250/EWG und Nr. 71/393/EWG der Kommission vom 15. Juni 1971 2 und vom 18. November 1971 3 haben bereits eine Reihe von Analysemethoden festgelegt. Nach dem Stand der seitdem durchgeführten Arbeiten ist es nunmehr geboten, eine dritte Serie von Analysemethoden festzulegen.
Die in dieser Richtlinie vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Futtermittelausschusses -
hat folgende Richtlinie erlassen:
Die Mitgliedstaaten schreiben vor, dass die Analysen für die amtliche Untersuchung von Futtermitteln auf ihren Gehalt an Stärke, Rohprotein, durch Pepsin und Salzsäure lösbarem Rohprotein und an freiem und Gesamtgossypol sowie auf die Pepsinaktivität nach den in Anlage I zu dieser Richtlinie beschriebenen Methoden durchgeführt werden.
Die Mitgliedstaaten schreiben vor, dass die Analysen für die amtliche Untersuchung von Futtermitteln auf den Gehalt der Futtermittel an Tylosin und Virginiamycin nach den in Anlage II zu dieser Richtlinie beschriebenen Methoden durchgeführt werden.
Die Mitgliedstaaten setzen spätestens zum 1. Juli 1973 die erforderlichen Rechts- oder Verwaltungsvorschriften in Kraft, um den Bestimmungen dieser Richtlinie nachzukommen. Sie setzen die Kommission unverzüglich hiervon in Kenntnis.
Diese Richtlinie ist an alle Mitgliedstaaten gerichtet.
| Anlage I |
1. BESTIMMUNG VON STÄRKE POLARIMETRISCHES VERFAHREN
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehaltes an Stärke und ihrer Abbauprodukte mit hohem Molekulargewicht in Futtermitteln zum Zweck der Prüfung ihrer Übereinstimmung mit der Richtlinie 86/174/EWG der Kommission und der Richtlinie 96/25/EG des Rates.
2. Prinzip
Die Methode basiert auf einer doppelten Bestimmung. Bei der ersten Bestimmung wird die Probe mit verdünnter Salzsäure heiß behandelt. Nach Klärung und Filtration wird die optische Drehung der Lösung polarimetrisch gemessen.
Bei der zweiten Bestimmung wird die Probe mit 40 % Äthanol extrahiert. Nach Behandlung des Filtrats mit Salzsäure wird geklärt, filtriert und die optische Drehung unter den gleichen Bedingungen wie bei der ersten Bestimmung gemessen.
Der Unterschied zwischen den beiden Messungen, multipliziert mit einem bekannten Faktor, ergibt den Stärkegehalt der Probe.
3. Reagenzien
3.1. 25 % (G/G) Salzsäure, D: 1,126.
3.2. 1,128 % Salzsäure (GN).
Die Konzentration muss durch Titration mit 0,1 N Natriumhydroxidlösung in Gegenwart von 0,1 % Methylrot (G/V) in 94 % Äthanol 94 (V/V) geprüft werden. 10 ml = 30,94 ml 0,1 N NaOH.
3.3. Carrez-Lösung I: 21,9 g Zinkazetat Zn (CH3COO )2:2H2O und 3 g Eisessig werden in Wasser gelöst und auf 100 ml mit Wasser aufgefüllt.
3.4. Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat (II), [K4(Fe (CN )6 *3H2O werden in Wasser gelöst und auf 100 ml mit Wasser aufgefüllt.
3.5. 40 % Äthanol (V/V), D: 0,948 bei 20 °C.
4. Geräte
4.1. 250-ml-Erlenmeyerkolben mit Schliffstopfen und Rückflußkühler.
4.2. Polarimeter oder Saccharimeter.
5. Ausführung
5.1. Vorbereitung der Probe
Die Probe muss so fein gemahlen werden, dass sie vollständig durch ein Rundlochsieb von 0,5 mm Lochdurchmesser hindurchgeht.
5.2. Bestimmung der gesamten optischen Drehung (P oder S) (siehe Bemerkung 7.1)
2,5 g der gemahlenen Probe werden auf 1 mg genau in einen 100-ml-Meßkolben eingewogen und 25 ml Salzsäure ( 3.2) hinzugefügt. Der Kolben wird geschüttelt, bis sich die Substanz gut verteilt hat; dann werden weitere 25 ml Salzsäure ( 3.2) hinzugegeben. Der Kolben wird in ein Bad mit kochendem Wasser gestellt und während der ersten 3 Minuten kräftig und regelmäßig umgeschüttelt, um die Bildung von Klumpen zu verhindern. Die in dem Wasserbad enthaltene Wassermenge muss ausreichen, um das Wasser auch dann noch über dem Siedepunkt zu halten, wenn der Kolben eingetaucht wird. Während des Schüttelns darf der Kolben nicht aus dem Wasser herausgenommen werden. Nach genau 15 Minuten wird der Kolben aus dem Wasserbad entfernt, 30 ml kaltes Wasser hinzugefügt und unverzüglich auf 20 °C abgekühlt.
Es werden 5 ml Carrez-Lösung I ( 3.3) hinzugefügt und 1 Minute geschüttelt; anschließend werden 5 ml Carrez-Lösung II ( 3.4) hinzugefügt und es wird nochmals 1 Minute geschüttelt. Dann wird bis zur Marke mit Wasser aufgefüllt, umgeschüttelt und filtriert. Ist das Filtrat nicht vollständig klar (was selten vorkommt), muss die Bestimmung mit größeren Mengen Carrez-Lösungen I und II, z.B. 10 ml, wiederholt werden.
Anschließend wird die optische Drehung der Lösung in einem 200-mm-Rohr mit einem Polarimeter oder einem Saccharimeter gemessen.
5.3. Bestimmung der optischen Drehung (P" oder S) der in Äthanol 40 v. H. löslichen Substanzen
5 g der Probe werden auf 1 mg genau in einen 100-ml-Meßkolben eingewogen und etwa 80 ml Äthanol ( 3.5) hinzugefügt (siehe Bemerkung 7.2). Anschließend wird der Kolben 1 Stunde lang bei Raumtemperatur stehengelassen und währenddessen sechsmal kräftig geschüttelt, damit sich die Substanz gut mit dem Äthanol vermischt. Es wird mit Äthanol ( 3.5) zur Marke aufgefüllt, umgeschüttelt und filtriert. 50 ml des Filtrats (= 2,5 g der Probe) werden in einen 250-ml-Erlenmeyerkolben abpipettiert und 2,1 ml Salzsäure ( 3.1) hinzugefügt; der Kolben wird kräftig geschüttelt, an einen Rückflußkühler angeschlossen und in ein Bad mit siedendem Wasser gesetzt. Nach genau 15 Minuten wird der Erlenmeyerkolben aus dem Wasserbad herausgenommen und der Inhalt in. einen 100-ml-Meßkolben mit einer kleinen Menge von kaltem Wasser überspült; anschließend wird auf eine Temperatur von 20 °C abgekühlt.
Darauf wird mit den Carrez-Lösungen I ( 3.3) und II ( 3.4) geklärt, mit Wasser zur Marke aufgefüllt, geschüttelt und filtriert und die optische Drehung gemessen, wie unter 5.2 zweiter und dritter Absatz beschrieben.
6. Berechnung der Ergebnisse
Der Stärkegehalt (%) wird wie folgt berechnet:
6.1. Polarimetrische Messungen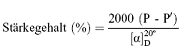
P = Optische Drehung insgesamt in Winkelgrad
P" = Optische Drehung in Winkelgrad der in Äthanol löslichen Substanzen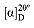 = Spezifische optische Drehung von reiner Stärke. Folgende numerische Werte sind allgemein anerkannt:
= Spezifische optische Drehung von reiner Stärke. Folgende numerische Werte sind allgemein anerkannt:
+ 185,9°: Reisstärke
+ 185,4°: Kartoffelstärke
+ 184,6°: Maisstärke
+ 182,7°: Weizenstärke
+ 181,5°: Gerstenstärke
+ 181,3°: Haferstärke
+ 184,0°: andere Stärkearten sowie Stärkegemische bei Mischfuttermitteln
6.2. Saccharimetrische Messungen
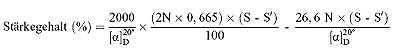
S = Optische Drehung insgesamt in Saccharimeter-Grad
S" = Optische Drehung in Saccharimeter-Grad der in Äthanol löslichen Substanzen
N = Gewicht (g) von Saccharose in100 ml Wasser, das bei Messung mit einem 200-mm-Rohr eine optische Drehung von 100 Saccharimeter-Grad bewirkt
16,29 g für die französischen Saccharimeter
26,00 g für die deutschen Saccharimeter.
20,00 g für gemischte Saccharimeter.= Spezifische optische Drehung von reiner Stärke (siehe 6.1)
6.3. Wiederholbarkeit
Der Unterschied zwischen zwei Parallelbestimmungen darf bei ein und derselben Probe bei Stärkegehalten von weniger als 40 % 0,4 % absolut und bei Gehalten von 40 % und mehr 1,1 % relativ nicht überschreiten.
7. Bemerkungen
7.1. Enthält die Probe mehr als 6 % Carbonate, berechnet als Calciumcarbonat, so müssen diese vor der Bestimmung der gesamten optischen Drehung durch Behandlung mit der genau notwendigen Menge verdünnter Schwefelsäure zerstört werden.
7.2. Bei Erzeugnissen mit hohem Laktosegehalt, z.B. bei Molkenpulver oder Magermilchpulver, wird nach Zusatz von 80 ml Äthanol ( 3.5) wie folgt verfahren: Der Meßkolben wird mit einem Rückflußkühler versehen und während 30 Minuten in ein Wasserbad von 50 °C getaucht. Nach Abkühlen wird das Verfahren wie unter 5.3 beschrieben fortgeführt.
7.3. Folgende Futtermittelbestandteile führen bei größeren Mengen in Futtermitteln erwiesenermaßen zu Interferenzen bei der Bestimmung des Stärkegehaltes durch das polarimetrische Verfahren und könnten so falsche Ergebnisse zur Folge haben:
2. BESTIMMUNG VON ROHPROTEIN
1. Zweck und Anwendungsbereich
Diese Methode dient der Bestimmung des Rohproteingehalts von Futtermitteln anhand des nach Kjeldahl ermittelten Stickstoffgehalts.
2. Prinzip
Die Probe wird mit Schwefelsäure in Anwesenheit eines Katalysators aufgeschlossen. Die saure Lösung wird mit Natronlauge alkalisiert. Das freigesetzte Ammoniak wird in eine Vorlage überdestilliert, die eine bestimmte Menge Schwefelsäure enthält, deren Überschuss mit einer Natronlauge-Standardlösung titriert wird.
3. Reagenzien
3.1 Kaliumsulfat.
3.2 Katalysator: Kupfer(II)-oxid CuO oder Kupfer(II)-sulfat-Pentahydrat CuSO4 ·5 H2O.
3.3 Zink, gekörnt.
3.4 Schwefelsäure, ρ20 = 1,84 g/ml.
3.5 Schwefelsäure, c (1/2 H2SO4) = 0,5 mol/l.
3.6 Schwefelsäure, c (1/2 H2SO4) = 0,1 mol/l.
3.7 Methylrot-Indikator: 300 mg Methylrot werden in 100 ml Ethanol (σ = 95 - 96 %) gelöst.
3.8 Natronlauge β = 40 g/100 ml (m/V: 40 %).
3.9 Natronlauge c = 0,25 mol/l.
3.10 Natronlauge c = 0,1 mol/l.
3.11 Bimsstein gekörnt, mit Salzsäure gewaschen und geglüht.
3.12 Acetanilid (Schmp. = 114 °C, N = 10,36 %).
3.13 Saccharose (stickstofffrei).
4. Geräte
Aufschluss-, Destillations- und Titrationsapparat nach Kjeldahl.
5. Ausführung
5.1 Aufschluss
1 g der Probe wird auf 0,001 g genau eingewogen und in den Kolben des Aufschlussapparats überführt. Es werden15 g Kaliumsulfat ( 3.1), eine geeignete Menge Katalysator ( 3.2) (0,3 bis 0,4 g Kupfer(II)-oxid oder 0,9 bis 1,2 g Kupfersulfat), 25 ml Schwefelsäure ( 3.4) und einige Bimssteinkörnchen ( 3.11) zugesetzt, und der Kolbeninhalt wird gemischt. Der Kolben wird, wenn notwendig, unter regelmäßigem Schwenken zunächst mäßig erhitzt, bis die Substanz verkohlt ist und das Schäumen aufgehört hat; sodann wird die Flüssigkeit stärker erhitzt und gleichmäßig am Sieden gehalten. Bei korrekter Erhitzung kondensiert die siedende Säure auf der Kolbenwand. Ein Überhitzen der Kolbenwände und Ansetzen organischer Partikel ist zu vermeiden. Sobald die Lösung klar ist und sich hellgrün färbt, wird sie noch weitere 2 Stunden am Sieden gehalten und danach abkühlen gelassen.
5.2 Destillation
Es wird vorsichtig soviel Wasser zugegeben, dass die Sulfate vollständig gelöst werden. Man lässt abkühlen; sodann werden einige Zinkkörnchen ( 3.3) zugesetzt.
In den Auffangkolben des Destillationsapparates werden je nach dem zu erwartenden Stickstoffgehalt genau 25 ml Schwefelsäure ( 3.5 oder 3.6) gebracht und einige Tropfen Methylrot-Indikator ( 3.7) hinzugefügt.
Der Aufschlusskolben wird mit dem Kühler des Destillationsapparats verbunden und das Ende des Kühlers mindestens 1 cm tief in die Flüssigkeit des Auffangkolbens gesenkt (siehe Bemerkung 8.3). Dann werden 100 ml Natronlauge ( 3.8) langsam in den Aufschlusskolben eingefüllt, wobei kein Ammoniak entweichen darf (siehe Bemerkung 8.1).
Der Kolben wird solange erhitzt bis alles Ammoniak überdestilliert ist.
5.3 Titration
Der Schwefelsäureüberschuß im Auffangkolben wird je nach Konzentration der verwendeten Schwefelsäure mit Natronlauge ( 3.9 oder 3.10) bis zum Endpunkt titriert.
5.4 Blindversuch
Zur Feststellung, ob die Reagenzien stickstofffrei sind, wird ein Blindversuch (Aufschluss, Destillation und Titration) mit 1 g Saccharose ( 3.13) anstelle der Probe durchgeführt.
6. Berechnung der Ergebnisse
Der Rohproteingehalt errechnet sich nach folgender Formel:
[(V0 - V1 ) * c * 0;014 * 100 * 6,25] / m
Hierinbedeuten:
V0 = Volumen (ml) der bei der Titration des Blindversuchs ( 5.4) verbrauchten Natronlauge ( 3.9 oder 3.10).
V1 = Volumen (ml) der bei der Titration ( 5.3) verbrauchten Natronlauge ( 3.9 oder 3.10).
c = Konzentration (mol/l) der Natronlauge ( 3.9 oder 3.10).
m = Masse der Probe in Gramm.
7. Zuverlässigkeit der Methode
7.1 Wiederholbarkeit
Der Unterschied der Ergebnisse von zwei Parallelbestimmungen darf bei ein und derselben Probe folgende Werte nicht überschreiten:
7.2 Richtigkeit
Die Analyse (Aufschluss, Destillation und Titration) wird mit 1,5 bis 2,0 g Acetanilid ( 3.12) in Gegenwart von 1 g Saccharose ( 3.13) durchgeführt; 1 g Acetanilid verbraucht 14,80 ml Schwefelsäure ( 3.5). Die Wiederfindung muss mindestens 99 % betragen.
8. Bemerkungen
8.1 Es können manuelle, halbautomatische oder automatische Geräte verwendet werden. Bei Geräten, die ein Umfüllen zwischen Aufschluss und Destillation erfordern, ist darauf zu achten, dass dies verlustlos geschieht. Verfügt der Destillationsapparat nicht über einen Tropftrichter, so erfolgt die Zugabe der Natronlauge unmittelbar vor dem Anschließen des Kolbens an den Kühler; die Flüssigkeit ist in diesem Fall langsam an den Kolbenwänden entlanglaufen zu lassen.
8.2 Kommt es während des Aufschlusses zur Verfestigung der Mischung, ist mit der Bestimmung von vorn zu beginnen und eine größere als die obengenannte Menge an Schwefelsäure ( 3.4) zu verwenden.
8.3 Bei stickstoffarmen Proben kann die in den Auffangkolben einzufüllende Menge Schwefelsäure ( 3.6) gegebenenfalls auf 10 oder 15 ml verringert und mit Wasser auf 25 ml aufgefüllt werden.
3. BESTIMMUNG DES DURCH PEPSIN UND SALZSÄURE LÖSBAREN ROHPROTEINS
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des durch Pepsin und Salzsäure lösbaren Rohproteins unter definierten Bedingungen. Sie gilt für alle Futtermittel.
2. Prinzip
Die Probe wird 48 Stunden lang bei 40° C mit einer Pepsin-Salzsäure-Lösung behandelt. Die Suspension wird filtriert und der Stickstoffgehalt des Filtrats nach der für die Bestimmung von Rohprotein beschriebenen Methode bestimmt.
3. Reagenzien
3.1 Salzsäure, D: 1,125
3.2 0,075 N Salzsäure
3.3 Pepsin 2.0 E/mg. Die Aktivität ist definiert in der Vorschrift des Teils 4 dieser Anlage und hiernach zu kontrollieren.
3.4 Pepsinlösung ca. 0,02 v.H. (G/V) in Salzsäure ( 3.2), frisch zubereitet. Aktivität 400 E/l
3.5 Antischaumemulsion (z.B. Silikon)
3.6 Sämtliche unter Punkt 3 der Methode zur Bestimmung von Rohproteinen aufgeführte Reagenzien
4. Geräte
4.1 Wasserbad oder Brutschrank, auf 40° C ± 1° C eingestellt
4.2 Apparatur zum Aufschluss und zur Destillation nach Kjeldahl
5. Ausführung
5.1 Lösen des Rohproteins (s. Bemerkung 7.2)
2 g der Probe werden auf 1 mg genau eingewogen und in einen 500-ml-Meßkolben gebracht. Dann fügt man 450 ml Pepsinlösung ( 3.4), die zuvor auf 40° C erwärmt wurden, hinzu und schüttelt um, um Klümpchenbildung zu verhindern. Es ist darauf zu achten, dass der pH-Wert der Suspension unter 1,7 liegt. Der Kolben wird ins Wasserbad oder in den Brutschrank ( 4.1) gestellt und dort 48 Stunden belassen. Nach 8, 24 und 32 Stunden ist er zu schütteln. Nach 48 Stunden werden 15 ml Salzsäure ( 3.1) zugesetzt, auf 20° C abkühlengelassen, mit Wasser zur Marke aufgefüllt und filtriert.
5.2 Aufschluss
250 ml des Filtrats werden abgenommen und in den Kolben der Aufschlussapparatur ( 4.2) gefüllt. Dann werden die zum Aufschluss benötigten Reagenzien, wie in der Beschreibung der Methode zur Bestimmung des Rohproteins unter Punkt 5.1, zweiter Satz, angegeben, hinzugefügt. Es wird gemischt und bis zum Sieden erhitzt. Falls sich starker Schaum bildet, werden einige Tropfen Antischaumemulsion ( 3.5) zugesetzt. Die Flüssigkeit wird im kräftigen Siedengehalten, bis das Wasser fast vollständig verdampft ist. Die letzten Wasserspuren werden vorsichtig bei verringerter Hitze beseitigt. Wenn die Lösung klar und farblos (bzw. hellgrün bei Verwendung eines kupferhaltigen Katalysators) ist, wird das Sieden noch eine Stunde lang fortgesetzt; dann lässt man abkühlen.
5.3 Destillation und Titration
Es ist wie in der Methode zur Bestimmung von Rohproteinen unter 5.2 und 5.3 angegeben zu verfahren.
5.4 Blindversuch
Ein Blindversuch wird unter Anwendung desselben Verfahrens ohne Analysenprobe ausgeführt.
6. Berechnung der Ergebnisse
Das Volumen an Schwefelsäure, das im Blindversuch verbraucht wird, wird von dem Verbrauch der Analysenprobe abgezogen. 1 ml 0,1 N Schwefelsäure entspricht 1,4 mg Stickstoff.
Die erhaltene Stickstoffmenge wird mit dem Faktor 6,25 multipliziert. Das Ergebnis wird in Hundertteilen der Probe ausgedrückt.
Wiederholbarkeit
Der Unterschied zwischen zwei Parallelbestimmungen darf bei ein und derselben Probe
7. Bemerkungen
7.1 Die ermittelten Werte stehen in keinem unmittelbaren Zusammenhang mit der Verdaulichkeit in vivo.
7.2 Futtermittel, deren Rohfettgehalt 10 v.H. übersteigt, sind durch Extraktion mit Petroläther (Kp 40 - 60° C) zu entfetten.
4. BESTIMMUNG DER PEPSINAKTIVITÄT
1. Zweck und Anwendungsbereich
Die Methode dient zur Kontrolle der Aktivität des Pepsins, das zur Bestimmung des durch Pepsin und Salzsäure lösbaren Rohproteins verwendet wird.
2. Prinzip
Hämoglobin wird unter definierten Bedingungen mit Pepsin und verdünnter Salzsäure behandelt. Der nicht hydrolysierte Anteil der Proteine wird mit Trichloressigsäure gefällt. Das Filtrat wird mit Natriumhydroxidlösung und mit Reagenz nach Folin-Ciocalteu versetzt. Die Extinktion dieser Lösung wird bei 750 nm gemessen und die dieser Extinktion entsprechende Menge Tyrosin einer Eichkurve entnommen.
Definition: Die Pepsin-Einheit (E) wird definiert als die Enzymmenge, die unter den Bedingungen der Methode pro Minute soviel Hydroxyaryl-Verbindungen freisetzt, dass deren Färbung mit Folin-Ciocalteu-Reagenz eine Extinktion ergibt, die derjenigen von 1 µ Mol Tyrosin unter den gleichen Bedingungen entspricht.
3. Reagenzien
3.1 0,2 N Salzsäure
3.2 0,06 N Salzsäure
3.3 0,025 N Salzsäure
3.4 Trichloressigsäurelösung 5 v.H. (G/V)
3.5 0,5 N Natriumhydroxidlösung
3.6 Reagenz nach Folin-Ciocalteu: 100 g Natriumwolframat (Na2WO4 · 2 H2O), 25 g Natriummolybdat (Na2MoO4 · 2 H2O) und 700 ml Wasser werden in einen 2-l-Rundkolben mit Schliffstopfen gegeben. Man fügt 50 ml Phosphorsäure (D: 1,71) und 100 ml konzentrierte Salzsäure (D: 1,19) hinzu, verbindet den Kolben mit einem Rückflußkühler, erhitzt zum Sieden und hält die Lösung 10 Stunden lang im gelinden Sieden. Nach dem Abkühlen entfernt man den Rückflußkühler, gibt 175 g Lithiumsulfat (Li2SO4 · 2 H2O), 50 ml Wasser und 1 ml Brom hinzu. Anschließend kocht man 15 Minuten lang zur Entfernung des überschüssigen Broms.
Nach Abkühlen überführt man die Lösung in einen 1-l-Meßkolben, füllt mit Wasser auf, schüttelt um und filtriert. Das fertige Reagenz darf keine grünliche Farbe aufweisen. Vor Gebrauch werden 1 Volumenteil Reagenz mit 2 Volumenteilen Wasser verdünnt.
3.7 Hämoglobinlösung: Eine 354 mg Stickstoff 4 entsprechende Hämoglobinmenge (ca. 2 g), Proteasesubstrat nach Anson, wird eingewogen und in einen 200-ml-Kolben mit Schliffstopfen gebracht. Man gibt einige ml 0,06 N Salzsäure ( 3.2) hinzu, schließt den Kolben an der Vakuumpumpe an und schüttelt, bis das Hämoglobin vollständig gelöst ist. Man entfernt das Vakuum und gibt unter Umschütteln die noch zu 100 ml fehlende Menge Salzsäure ( 3.2) hinzu. Vor Gebrauch frisch herzustellen.
3.8 Tyrosin-Standardlösung: 181,2 mg Tyrosin werden in Salzsäure ( 3.1) gelöst und zu 1 l mit derselben Säure aufgefüllt (Stammlösung). 20,0 ml dieser Lösung werden entnommen und mit Salzsäure ( 3.1) auf 100 ml verdünnt. 1 ml dieser Lösung enthält 0,2 µ Mol Tyrosin.
4. Geräte
4.1 Ultrathermostat, auf 25° C ± 0,1° C eingestellt
4.2 Spektralphotometer
4.3 Chronometer, Genauigkeit: 1 sec.
4.4 pH-Meßgerät
5. Ausführung
5.1 Herstellung der Lösung (s. Bemerkung 7.1)
150 mg Pepsinwerdenin100 ml Salzsäure ( 3.2) gelöst. Von dieser Lösung werden 2 ml abpipettiert und in einem 50-ml-Meßkolben mit Salzsäure ( 3.3) zur Marke aufgefüllt. Der mit dem pH-Meßgerät kontrollierte pH-Wert sollte 1,6 ± 0,1 betragen. Der Kolben wird in den Ultrathermostaten ( 4.1) gebracht.
5.2 Hydrolyse
5,0 ml Hämoglobinlösung ( 3.7) werden in ein Reagenzglas pipettiert und im Ultrathermostat ( 4.1) auf 25° C erwärmt. Dann gibt man 1,0 ml der nach 5.1 erhaltenen Pepsinlösung zu, mischt durch etwa 10maliges Auf- und Abbewegen eines am Ende verdickten Glasstabes und belässt das Reagenzglas, von der Zugabe der Pepsinlösung an gerechnet, genau 10 Minuten lang bei 25° C im Wasserbad (Zeit und Temperatur sind genau einzuhalten). Nach Ablauf dieser Zeit fügt man 10,0 ml der zuvor auf 25° C gebrachten Trichloressigsäurelösung ( 3.4) hinzu, schüttelt um und filtriert durch eintrockenes Filter.
5.3 Entwicklung der Färbung und Messung der Extinktion
5,0 ml des Filtrats werden in einen 50-ml-Erlenmeyerkolben pipettiert, mit 10,0 ml Natriumhydroxidlösung ( 3.5) versetzt, dann werden unter stetigem Umschütteln 3,0 ml des verdünnten Reagenz nach Folin-Ciocalteu ( 3.6) hinzugefügt. Nach 5 bis 10 Minuten wird die Extinktion der Lösung in 1-cm-Küvetten bei 750 nm gegen Wasser photometrisch bestimmt.
5.4 Blindversuch
Zu jeder Bestimmung ist ein Blindversuch wie folgt durchzuführen:
5,0 ml Hämoglobinlösung ( 3.7) werden in ein Reagenzglas pipettiert und im Ultrathermostaten ( 4.1) auf 25° C erwärmt. Dann gibt man10,0 ml der zuvor auf 25° C gebrachten Trichloressigsäurelösung ( 3.4) hinzu, mischt und fügt anschließend 1,0 ml der nach 5.1 erhaltenen Pepsinlösung hinzu. Man mischt mittels eines Glasstabes und belässt das Reagenzglas genau 10 Minuten lang bei 25° C im Wasserbad ( 4.1). Es wird umgeschüttelt und filtriert. Dann wird das Verfahren wie unter 5.3 beschrieben fortgeführt.
5.5 Eichkurve
In 50-ml-Erlenmeyerkolben werden 1,0, 2,0, 3,0, 4,0 und 5,0 ml Tyrosinstandardlösung ( 3.8) (entsprechend den Tyrosinmengen von 0,2, 0,4, 0,6, 0,8 und 1,0 µ Mol) gegeben. Die Reihe wird durch eine tyrosinfreie Vergleichsprobe vervollständigt. Die einzelnen Ansätze werden mit Salzsäure ( 3.1) auf 5,0 ml ergänzt. Weiter werden 10,0 ml Natriumhydroxidlösung ( 3.5) und unter ständigem Schütteln 3,0 ml des verdünnten Reagenz nach Folin-Ciocalteu ( 3.6) zugegeben. Die Extinktionwird gemäß 5.3, letzter Satz, gemessen. Die Eichkurve wird durch Auftragen der zugesetzten Mengen Tyrosin gegen die Extinktion festgelegt.
6. Berechnung der Ergebnisse
Man entnimmt der Eichkurve die Menge an Tyrosin in µ Mol, die der durch den Blindwert korrigierten Extinktion der gefärbten Lösungen entspricht.
Die Aktivität des Pepsins in µ Mol Tyrosin pro mg und pro Minute bei 25° C ist nach folgender Formel zu berechnen:
Einheiten pro mg (E/mg ) = 0,32 a / p
wobei:
a = aus der Eichkurve entnommene Tyrosinmenge in µ Mol,
p = Gewicht in mg der gemäß 5.2 zugesetzten Pepsinmenge.
7. Bemerkung
7.1 Die aufzulösende Menge an Pepsin ist so zu wählen, dass bei der photometrischen Endmessung eine Extinktion von 0,35 ± 0,035 erreicht wird.
7.2 2 E/mg, bestimmt nach dieser Methode, entsprechen
3,64 m-Anson E/mg (µ Mol Tyrosin/mg · Min. bei 35,5° C) oder
36.400 handelsüblichen E/g (µ Mol Tyrosin/g · 10 Min. bei 35,5° C).
5. BESTIMMUNG VON FREIEM UND GESAMTGOSSYPOL
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung von freiem Gossypol, Gesamtgossypol und chemisch nahe verwandter Substanzen in Samen, Mehl und Kuchen von Baumwollsaat sowie in Mischfuttermitteln, die Erzeugnisse aus Baumwollsaat enthalten. Die untere Grenze der Bestimmbarkeit beträgt 20 ppm.
2. Prinzip
Gossypol wird in Gegenwart von 3-Amino-1-Propanol entweder mit einer Isopropanol-Hexan-Mischung für die Bestimmung von freiem Gossypol oder mit Dimethylformamid für die Bestimmung von Gesamtgossypol extrahiert und mittels Anilin zu Gossypoldianilin umgesetzt, dessen Extinktion bei 440 nm gemessen wird.
3. Reagenzien
3.1 Isopropanol-Hexan-Mischung: 60 Volumenteile Isopropanol p.a. werden mit 40 Volumenteilen n-Hexan gemischt.
3.2 Lösungsmittel A: In einen 1-l-Meßkolben werden ungefähr 500 ml Isopropanol-Hexan-Mischung ( 3.1), 2 ml 3-Amino-1-Propanol, 8 ml Eisessig und 50 ml Wasser gefüllt und mit Isopropanol-Hexan-Mischung ( 3.1) zur Marke aufgefüllt. Dieses Reagenz ist eine Woche lang haltbar.
3.3 Lösungsmittel B: In einen 100-ml-Meßkolben werden 2 ml 3-Amino-1-Propanol und 10 ml Eisessig abpipettiert, auf Raumtemperatur abgekühlt und mit N,N-Dimethylformamid zur Marke aufgefüllt. Dieses Reagenz ist eine Woche lang haltbar.
3.4 Anilin p.a.: Wenn die Extinktion im Blindversuch 0,022 überschreitet, ist das Anilin über Zinkstaub unter Verwerfung der ersten und letzten 10 v.H. Teile des Destillats zu destillieren. In einem braunen, geschlossenen Gefäß ist dieses Reagenz einige Monate im Kühlschrank haltbar.
3.5 Gossypol-Standardlösung A: In einem 250-ml-Meßkolben werden 27,9 mg Gossypol-Acetat mit Lösungsmittel a ( 3.2) gelöst und dann damit zur Marke aufgefüllt. 50 ml dieser Lösung werden in einen 250-ml-Meßkolben pipettiert und mit Lösungsmittel a ( 3.2) zur Marke aufgefüllt. Die Konzentration dieser Lösung an Gossypol beträgt 0,02 mg/ml. Vor Gebrauch lässt man 1 Stunde lang bei Raumtemperatur stehen.
3.6 Gossypol-Standardlösung B: In einem 50-ml-Meßkolben werden 27,9 mg Gossypol-Acetat in Lösungsmittel B ( 3.3) gelöst und zur Marke aufgefüllt. Die Konzentration dieser Lösung an Gossypol beträgt 0,5 mg/ml.
Die Standard-Gossypol-Lösungen sind vor Lichteinwirkung geschützt 24 Stunden lang haltbar.
4. Geräte
4.1 Mechanischer Schüttelapparat: ungefähr 35 U/min.
4.2 Spektralphotometer.
5. Ausführung
5.1 Einwaage
Die Größe der Einwaage richtet sich nach dem vermuteten Gehalt an Gossypol in der Probe. Es ist vorteilhaft, mit einer geringen Einwaage und einem relativ großen aliquoten Teil des Filtrats zu arbeiten, um genügend Gossypol für eine genaue photometrische Messung zu erhalten. Für die Bestimmung von freiem Gossypol in Samen, Mehl und Kuchen von Baumwollsaat soll die Einwaage 1 g nicht überschreiten, während sie bei Mischfuttermitteln 5 g erreichen kann. Ein aliquoter Teil des Filtrats von 10 ml ist in den meisten Fällen anwendbar, er soll 50 bis 100 µg Gossypol enthalten. Für die Bestimmung von Gesamtgossypol soll die Einwaage 0,5 bis 5 g betragen, so dass im aliquoten Teil des Filtrats von 2 ml 40 bis 200 µg Gossypol enthalten sind.
Die Analyse ist bei einer Raumtemperatur nahe 20° C auszuführen.
5.2 Bestimmung von freiem Gossypol
Die Einwaage wird in einen 250-ml-Kolben mit Schliffstopfen, dessen Boden mit Glassplittern bedeckt ist, übergeführt, 50 ml des Lösungsmittels a ( 3.2) werden hinzupipettiert, der Kolben wird verschlossen und 1 Stunde lang im Schüttelapparat bewegt. Dann wird durch ein trockenes Filter filtriert und das Filtrat in einem kleinen Kolben mit Schliffstopfen aufgefangen. Währen der Filtration wird der Trichter mit einem Uhrglas bedeckt. Es werden gleich große aliquote Teile des Filtrats, die 50 bis 100 µg Gossypol enthalten, in zwei 25-ml-Meßkolben (a und B) pipettiert, gegebenenfalls wird auf 10 ml mit Lösungsmittel a ( 3.2) ergänzt.
Dann wird der Inhalt des Kolbens (A) mit der Isopropanol-Hexan-Mischung ( 3.1) zur Marke aufgefüllt. Diese Lösung wird als Vergleichslösung für die Messung der Probelösung verwendet.
Danach werden in zwei andere 25-ml-Meßkolben (C und D) 10 ml Lösungsmittel a ( 3.2) pipettiert. Der Inhalt des Kolbens (C) wird mit der Isopropanol-Hexan-Mischung ( 3.1) zur Marke aufgefüllt. Diese Lösung wird als Vergleichslösung für die Messung der Blindprobelösung verwendet.
Zu den Meßkolben (D) und (B) werden je 2 ml Anilin ( 3.4) hinzugefügt. Es wird 30 Minuten lang auf einem Bad mit kochendem Wasser zur Entwicklung der Färbung erhitzt, auf Raumtemperatur abgekühlt, mit der Isopropanol-Hexan-Mischung ( 3.1) zur Marke aufgefüllt, geschüttelt und 1 Stunde lang stehen gelassen.
Dann werden im Spektralphotometer bei 440 nm und in Glasküvetten von 1 cm Dicke die Extinktion der Blindprobe (D) im Vergleich mit der Vergleichslösung (C) und die Extinktion der Probelösung (B) im Vergleich mit der Vergleichslösung (A) gemessen. Die Extinktion der Blindprobelösung wird von der Extinktion der Probelösung abgezogen (= korrigierte Extinktion). Aus dem erhaltenen Wert wird der Gehalt an freiem Gossypol wie unter 6 angegeben berechnet.
5.3 Bestimmung von Gesamtgossypol
Eine Einwaage, die 1 bis 5 mg Gossypol enthält, wird in einen 50-ml-Meßkolben gegeben; dann werden 10 ml des Lösungsmittels B ( 3.3) hinzugefügt. Gleichzeitig wird ein Blindversuch mit 10 ml des Lösungsmittels B ( 3.3) in einen anderen 50-ml-Meßkolben vorbereitet. Beide Meßkolben werden 30 Minuten lang auf einem Bad mit kochendem Wasser erhitzt, bei Raumtemperatur abkühlengelassenund mit der Isopropanol-Hexan-Mischung ( 3.1) zur Marke aufgefüllt. Es wird geschüttelt, 10 bis 15 Minuten stehen gelassen, dann filtriert und das Filtrat in Kolben mit Schliffstopfen aufgefangen.
Es werden je 2 ml des Filtrats der Probe in zwei 25-ml-Meßkolben und je 2 ml des Filtrats des Blindversuchs in zwei andere 25-ml-Meßkolben pipettiert. Je ein Kolben jeder Serie wird auf 25 ml mit der Isopropanol-Hexan-Mischung ( 3.1) aufgefüllt. Diese Lösungen werden als Vergleichslösungen verwendet.
Zu den beiden anderen Meßkolben werden je 2 ml Anilin ( 3.4) hinzugefügt. Dann wird 30 Minuten lang auf einem Bad mit kochendem Wasser zur Entwicklung der Färbung erhitzt, auf Raumtemperatur abgekühlt, auf 25 ml mit der Isopropanol-Hexan-Mischung ( 3.1) aufgefüllt, geschüttelt und 1 Stunde lang stehen gelassen.
Die Extinktionen werden wie unter 5.2 für freies Gossypol angegeben gemessen. Aus dem ermittelten Wert wird der Gehalt an Gesamtgosspyol wie unter 6 angegeben berechnet.
6. Berechnung der Ergebnisse
Die Berechnung der Ergebnisse kann aus der spezifischen Extinktion ( 6.1) oder unter Benutzung einer Eichkurve ( 6.2) vorgenommen werden.
6.1 Aus der spezifischen Extinktion
Die spezifische Extinktion beträgt unter den angegebenen Bedingungen für
freies Gossypol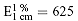
Gesamtgossypol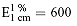
Der Gehalt an freiem oder Gesamtgossypol der Probe beträgt:
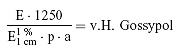
wobei:
E = korrigierte Extinktion, wie unter 5.2 bestimmt,
p = Einwaage in g,
a = aliquoter Teil des Filtrats in ml.
6.2 Mit Hilfe einer Eichkurve
6.2.1 Freies Gossypol
Es werden 2 Reihen von je fünf 25-ml-Meßkolbenvorbereitet. In beide Reihen werden 2,0, 4,0, 6,0, 8,0 und 10,0 ml der Gossypol-Standardlösung a ( 3.5) pipettiert und das Volumen auf 10 ml mit dem Lösungsmittel a ( 3.2) ergänzt. Jeder Reihe wird ein 25-ml-Meßkolben beigegeben, der nur 10 ml des Lösungsmittels a ( 3.2) enthält (Blindversuch). Die Kolben der ersten Reihe einschließlich des Blindversuchs werden mit der Isopropanol-Hexan-Mischung ( 3.1) auf 25 ml aufgefüllt (Vergleichsreihe).
Zu den Kolben der zweiten Reihe, einschließlich des Blindversuchs, werden je 2 ml Anilin ( 3.4) hinzugefügt. Dann wird 30 Minuten lang auf einem Bad mit kochendem Wasser zur Entwicklung der Färbung erhitzt, auf Raumtemperatur abgekühlt, zur Marke mit der Isopropanol-Hexan-Mischung ( 3.1) aufgefüllt, geschüttelt und 1 Stunde lang stehen gelassen (Standardreihe).
Unter den unter 5.2 angegebenen Bedingungen wird die Extinktion der Standardreihelösungen durch Vergleich mit den entsprechenden Lösungen der Vergleichsreihe gemessen. Die Eichkurve wird graphisch dargestellt, indem die Extinktionswerte und Gossypolmengen (in µg) gegeneinander eingetragen werden.
6.2.2 Gesamtgossypol
Es werden sechs 50-ml-Meßkolbenvorbereitet. In den ersten werden 10 ml des Lösungsmittels B ( 3.3) und in die übrigen je 2,0, 4,0, 6,0, 8,0 und 10,0 ml der Gossypolstandardlösung B ( 3.6) pipettiert. Der Inhalt jedes Kolbens wird auf 10 ml mit dem Lösungsmittel B ( 3.3) ergänzt, 30 Minuten lang auf einem Bad mit kochendem Wasser erhitzt, auf Raumtemperatur abgekühlt, mit der Isopropanol-Hexan-Mischung ( 3.1) zur Marke aufgefüllt und geschüttelt.
In zwei Reihen von sechs 25-ml-Meßkolbenwerdenje 2,0 ml dieser Lösung pipettiert. Die Kolben der ersten Reihe werden mit der Isopropanol-Hexan-Mischung ( 3.1) auf 25 ml aufgefüllt (Vergleichsreihe).
In die Kolben der zweiten Reihe werden je 2 ml Anilin ( 3.4) hinzugefügt. Dann werden sie 30 Minuten lang auf einem Bad mit kochendem Wasser erhitzt, auf Raumtemperatur abgekühlt, mit der Isopropanol-Hexan-Mischung ( 3.1) zur Marke aufgefüllt, geschüttelt und 1 Stunde lang stehen gelassen (Standardreihe).
Unter den unter 5.2 angegebenen Bedingungen wird die Extinktion der Standardreihelösungen durch Vergleich mit den entsprechenden Lösungen der Vergleichsreihe gemessen. Die Eichkurve wird graphisch dargestellt, indem die Extinktionswerte und Gossypolmengen (in µg) gegeneinander eingetragen werden.
6.3 Wiederholbarkeit
Der Unterschied zwischen zwei Parallelbestimmungen darf bei ein und derselben Probe bei Gehalten von
weniger als 500 ppm Gossypol 15 v.H. relativ,
nicht überschreiten.
| Anlage II |
4. BESTIMMUNG VON TYLOSIN
- durch Diffusion in Agarnährboden-
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung von Tylosin in Futtermitteln, Konzentrationen und Vormischungen. Die untere Grenze der Bestimmbarkeit beträgt 2 ppm.
2. Prinzip
Die Probe wird mit einer auf 80° C erwärmten Phosphatpufferlösung pH 8 behandelt und danach mit Methanol extrahiert. Nach Zentrifugieren wird der Extrakt verdünnt und seine antibiotische Aktivität durch Messung der Diffusion des Tylosins in einen mit Sarcina lutea beimpften Agarnährboden bestimmt. Die Diffusion wird durch die entstehenden Hemmzonen mit dem Testorganismus nachgewiesen. Der Durchmesser dieser Hemmzonen ist dem Logarithmus der Konzentration direkt proportional.
3. Testorganismus:
Sarcina lutea ATCC Nr. 9341
3.1 Haltung der Stammkultur
Ein Agarschrägröhrchen aus dem Nährboden ( 4.1), auf pH 7,0 eingestellt, wird mit Sarcina lutea beimpft und über Nacht bei etwa 35° C bebrütet. Danach wird die Kultur im Kühlschrank aufbewahrt und monatlich in Agarschrägröhrchen überimpft.
3.2 Herstellung der Keimsuspension
Ein Agarschrägröhrchen ( 3.1) wird mit 2 bis 3 ml steriler, frisch hergestellter Kochsalzlösung ( 4.4) aufgenommen. Mit dieser Suspension wird eine Rouxflasche, die 250 ml des Nährbodens ( 4.1) auf pH 7,0 enthält, beimpft. Nach einer Inkubationszeit von 24 Stunden bei 35° C werden die Keime mit 25 ml physiologischer Kochsalzlösung ( 4.4) aufgenommen und homogenisiert. Diese Suspension wird verdünnt, um eine Lichtdurchlässigkeit bei 650 nm von etwa 75 v.H. zu erhalten. Diese Suspension kann während einer Woche verwendet werden, wenn sie im Kühlschrank aufbewahrt wird.
Durch Versuchsplatten mit dem Testnährboden ( 4.1) wird die Impfmenge ermittelt, die möglichst große, aber noch scharfe Hemmzonen mit den verschiedenen verwendeten Konzentrationen an Tylosin ergibt. Die Beimpfung des Nährbodens muss zwischen 48 und 50° C erfolgen.
4. Nährboden und Reagenzien
Glukose 1 g
Pepton, tryptisch verdaut 10 g
Fleischextrakt 1,5 g
Hefeextrakt 3 g
Agar je nach Qualität 10 bis 20 g
Destilliertes Wasser ad 1000 ml
Für die Haltung der Stammkultur und Herstellung der Keimsuspension auf pH 7,0. für die Bestimmung auf pH 8,0 einstellen.
4.2 Phosphatpuffer, pH 8
Monokaliumphosphat KH2PO4 p.a. 0,523 g
Dikaliumphosphat K2HPO4 p.a. 16,730 g
Destilliertes Wasser ad 1000 ml
4.3 Phosphatpuffer, pH 7
Monokaliumphosphat KH2PO4 p.a. 5,5 g
Dikaliumphosphat K2HPO4 p.a. 13,6 g
Destilliertes Wasser ad 1000 ml
4.4 Physiologische Kochsalzlösung, steril
4.5 Methanol, rein
4.6 Methanol 40 v.H. (V/V)
4.7 Gemisch von Phosphatpuffer ( 4.2) und Methanol, rein: 60/40 in Vol.
4.8 Standardsubstanz: Tylosin von bekannter Aktivität.
5. Standardlösungen
Die Standardsubstanz ( 4.8) wird 3 Stunden lang bei 60° C im Vakuumtrockenschrank bei 5 mm Quecksilber getrocknet. Eine Einwaage von 10 bis 50 mg wird mit 5 ml Methanol ( 4.5) in einem Messkolben gelöst und dann mit Phosphatpuffer pH 7 ( 4.3) verdünnt, um eine Konzentration an Tylosinbase von1000 µg/ml zu erhalten.
Aus dieser Stammlösung wird durch Verdünnung mit dem Gemisch ( 4.7) eine Arbeitsstandardlösung S8 hergestellt, die 2 µg Tylosinbase/ml enthält. Dann werden die nachstehenden Konzentrationen mit dem Gemisch ( 4.7) durch aufeinanderfolgende Verdünnungen (1 + 1) hergestellt:
| S4 | 1 µg/ml |
| S2 | 0,5 µg/ml |
| S1 | 0,25 µg/ml |
6. Extraktion
Bei Konzentraten werden 10 g eingewogen, bei Vormischungen und Mischfuttermitteln 20 g. Die Einwaage wird mit 60 ml auf 80° C erwärmtem Phosphatpuffer pH 8 ( 4.2) versetzt und während 2 Minuten homogenisiert (Küchenmaschine, Ultra-Turrax etc.).
Danach lässt man 10 Minuten stehen, versetzt mit 40 ml Methanol ( 4.5) und homogenisiert erneut während 5 Minuten. Der Extrakt wird zentrifugiert und ein aliquoter Teil mit dem Gemisch ( 4.7) verdünnt, um eine vermutete Konzentration an Tylosin von 2 µg/ml (= U8) zu erhalten. Anschließend werden die Konzentrationen U4, U2 und U1 mit dem Gemisch ( 4.7) durch aufeinanderfolgende Verdünnungen (1 + 1) hergestellt.
Bei Gehalten unter 10 ppm wird der Extrakt bei 35° C zur Trockne auf einem Rotationsverdampfer eingeengt und mit Methanol 40 v.H. (4.6) aufgenommen.
7. Testverfahren
7.1 Beimpfung des Nährbodens
Der auf pH 8,0 eingestellte Testnährboden ( 4.1) wird mit der Keimsuspension ( 3.2) bei 48 bis 50° C beimpft.
7.2 Herstellung der Platten
Der Test ist als Diffusionstest auf Platten mit je 4 Konzentrationsstufen der Standardlösung (S8, S4, S2, S1) und des Extrakts (U8, U4, U2, U1) angelegt. Jede Platte erhält unbedingt alle 4 Konzentrationen des Standards und der Probe.
Daher muss die Plattengröße so bemessen sein, dass mindestens 8 Löcher in der Nährbodenschicht ausgestanzt werden können. Der Lochdurchmesser beträgt 10 bis 13 mm. Die beimpfte Nährbodenmenge ( 7.1) soll so berechnet sein, dass man eine gleichmäßige Schicht von ca. 2 mm Höhe erhält. Der Test sollte vorzugsweise auf Platten durchgeführt werden, die aus einer Glasscheibe mit einem darauf gelegten plangedrehten Aluminium- oder Kunststoffring von 20 mm Höhe und 200 mm Durchmesser hergestellt werden.
In die Löcher werden mit der Pipette genau abgemessene Mengen der Antibiotikalösung, zwischen 0,10 und 0,15 ml je nach Durchmesser der Löcher, gefüllt.
Für jede Probe muss mindestens jede Konzentration in 4facher Wiederholung eingefüllt werden, so dass pro Ansatz für jede Probe 32 Hemmzonen ausgewertet werden.
7.3 Inkubation
Die Inkubation erfolgt über Nacht bei 35 bis 37° C.
8. Auswertung
Der Durchmesser der Hemmzonen wird vorzugsweise durch Projektion gemessen. Die Messungen werden auf Semilogarithmenpapier aufgetragen, wobei der Logarithmus der Konzentration gegen den Durchmesser der Hemmzonen angetragen wird. Für den Standard sowie für den Extrakt werden die Geraden gezogen. In einem störungsfreien Test müssen die beiden Geraden parallel sein.
Der Logarithmus der relativen Aktivität wird durch folgende Formel rein rechnerisch ermittelt:

Tatsächliche Aktivität = angenommene Aktivität × relative Aktivität.
9. Wiederholbarkeit
Der Unterschied zwischen zwei Parallelbestimmungen darf bei ein und derselben Probe 10 v.H. relativ nicht überschreiten.
5. BESTIMMUNG VON VIRGINIAMYCIN
- durch Diffusion in Agarnährboden-
1. Zweck und Anwendungsbereich
Die Methode dient zur Bestimmung von Virginiamycin in Futtermitteln und Vormischungen. Die untere Grenze der Bestimmbarkeit beträgt 2 mg/kg (2 ppm) 6
2. Prinzip
Die Probe wird mit einer methanolischen Tween-80-Lösung extrahiert. Der Extrakt wird dekantiert oder zentrifugiert und verdünnt. Seine antibiotische Aktivität wird durch Messung der Diffusion des Virginiamycin in einem mit Micrococcusluteus beimpften Agarnährboden bestimmt. Die Diffusion wird durch die entstehenden Hemmzonen des Testorganismus nachgewiesen. Der Durchmesser dieser Hemmzonen wird für den Bereich der angewandten Konzentrationen als dem Logarithmus der Konzentration an Antibiotikum direkt proportional angesehen.
3. Testorganismus:
Micrococcus luteus ATCC 9341 (NCTC 8340, NCIB 8553)
3.1. Haltung der Stammkultur
Agarschrägröhrchen mit Kulturnährboden ( 4.1) werden mit Micrococcus luteus beimpft und 24 Stunden lang bei 30 °C bebrütet. Die Kultur wird im Kühlschrank bei etwa 4 °C aufbewahrt und alle zwei Wochen neu überimpft.
3.2. Herstellung der Bakteriensuspension 7
Eine junge Kultur auf Agarschrägröhrchen ( 3.1) wird mit 2 bis 3 ml Natriumchloridlösung ( 4.3) abgewaschen. Mit dieser Suspension wird eine Rouxflasche, die 250 ml des Nährbodens ( 4.1) enthält, beimpft und 18-20 Stunden lang bei 30 °C bebrütet. Die Kultur wird in 25 ml Natriumchloridlösung ( 4.3) aufgenommen und gemischt. Die Suspension wird 1: 10 mit Natriumchloridlösung ( 4.3) verdünnt. Die Lichtdurchlässigkeit der Suspension soll bei 650 nm und 1 cm Schichtdichte gegen Natriumchloridlösung ( 4.3) bei 75 v. H. liegen. Diese Suspension kann eine Woche lang bei ungefähr 4 °C aufbewahrt werden.
4. Nährboden und Reagenzien
4.1 Kultur- und Testnährboden 8
| Fleischpepton | 6,0 g, |
| Caseinpepton | 4,0 g, |
| Hefeextrakt | 3,0 g, |
| Fleischextrakt | 1,5 g, |
| Glukose | 1,0 g, |
| Agar | 10,0 bis 20,0 g, |
| Wasser pH 6,5 (nach Sterilisation) |
1 000 ml, |
4.2. Phosphatpuffer, pH 6
| Di-Kaliumhydrogenphosphat, K2HPO4 | 2,0 g, |
| Kaliumdihydrogenphosphat, KH2PO4 | 8,0 g, |
| Wasser | 1.000 ml. |
4.3. Natriumchloridlösung 0,8 v. H. (G/V): 8 g Natriumchlorid werden in Wasser aufgelöst, auf 1.000 ml verdünnt und sterilisiert.
4.4. Methanol.
4.5. Mischung des Phosphatpuffers ( 4.2) / Methanol ( 4.4): 80/20 (V/V).
4.6. Methanolische Tween-80-Lösung 0,5 v. H. (G/V): 5 g Tween 80 werden in Methanol ( 4.4) aufgelöst und auf 1.000 ml mit Methanol verdünnt.
4.7. Standardsubstanz: Virginiamycin mit bekannter Aktivität.
5. Standardlösungen
Eine genau abgewogene Menge der Standardsubstanz ( 4.7) wird in Methanol ( 4.4) gelöst und verdünnt um eine Stammlösung zu erhalten, die 1 000 µg Virginiamycin je ml enthält.
In verschlossener Flasche bei 4 °C aufbewahrt hält sich diese Lösung 5 Tage lang.
Aus dieser Stammlösung werden durch aufeinanderfolgende Verdünnungen mit der Mischung ( 4.5) folgende Lösungen hergestellt:
| S8 | 1 | µg/ml, |
| S4 | 0,5 | µg/ml, |
| S2 | 0,25 | µg/ml, |
| S1 | 0,125 | µg/ml. |
6. Herstellung des Extrakts und der Testlösungen
6.1. Extraktion
6.1.1. Erzeugnisse mit einem Virginiamycingehalt bis zu 100 mg/kg
Eine Menge von 50,0 g der Probe wird abgewogen, mit 200 ml der Lösung ( 4.6) versetzt und 30 Minuten geschüttelt. Nach dem Absetzen oder Zentrifugieren werden 20 ml des Überstands entnommen und in einem Vakuum-Rotationsverdampfer bei einer Temperatur nicht höher als 40 °C auf ca. 5 ml verdampft. Der Rückstand wird mit der Mischung ( 4.5) verdünnt, um einen erwarteten Virginiamycingehalt von 1 µg/ml (= U8) zu erhalten.
6.1.2. Erzeugnisse mit einem Virginiamycingehalt über 100 mg/kg
Eine Menge von höchstens 10,0 g der Probe mit einem Virginiamycingehalt zwischen 1 und 50 mg wird abgewogen, mit 100 ml der Lösung ( 4.6) versetzt und 30 Minuten lang geschüttelt. Nach dem Absetzen oder Zentrifugieren wird der Überstand mit der Mischung ( 4.5) verdünnt, um einen erwarteten Virginiamycingehalt von1 µg/ml (= U8) zu erhalten.
6.2. Testlösungen
Von der Lösung U8 werden die Lösungen U4 (erwarteter Gehalt: 0,5 µg/ ml), U2 (erwarteter Gehalt: 0,25 µg/ml) und U1 (erwarteter Gehalt: 0,125 µg/ml) durch aufeinanderfolgende Verdünnung (1: 1) mit der Mischung ( 4.5) hergestellt.
7. Testverfahren
7.1. Beimpfung des Testnährbodens
Der Testnährboden ( 4.1) wird mit der Bakteriensuspension ( 3.2) bei ungefähr 50 °C beimpft. Bei Vorversuchen auf Platten mit dem Testnährboden ( 4.1) wird die Menge des Inokulums ermittelt, die möglichst große und scharfe Hemmzonen mit den verschiedenen Virginiamycin-Konzentrationen ergibt.
7.2. Herstellung der Platten
Der Test ist als Diffusionstest auf Platten mit je vier Konzentrationsstufen der Standardlösung (S8, S4, S2, S1) und der Testlösung (U8, U4, U2, U1) angelegt. Jede Platte erhält unbedingt alle vier Konzentrationen des Standards und des Extrakts. Daher muss die Plattengröße so bemessen sein, dass mindestens acht Löcher mit einem Durchmesser von 10-13 mm und mindestens 30 mm zwischen den Lochzentren in die Nährbodenschicht gestanzt werden können. Der Test sollte vorzugsweise auf Platten durchgeführt werden, die aus einer Glasscheibe mit einem daraufgelegten plangedrehten Aluminium- oder Kunstoffring von 20 mm Höhe und 200 mm Durchmesser hergestellt werden. Auf die Platten wird eine Menge des nach 7.1 beimpften Testnährbodens ( 4.1) gegossen, so dass sich eine ca. 2 mm dicke Schicht ergibt (60 ml für eine Platte von 200 mm Durchmesser). Nachdem die Schicht festgeworden ist, werden die Löcher gestanzt und genau abgemessene Mengen der Standard- und Testlösungen einpipettiert. (Zwischen 0,10 und 0,15 ml pro Loch, je nach Lochdurchmesser). Jede Konzentration muss mindestens in vierfacher Wiederholung angewendet werden, so dass für jeden Test 32 Hemmzonen ausgewertet werden.
7.3. Inkubation
Die Inkubation erfolgt während 16 bis 18 Stunden bei 30 °C ± 2 °C.
8. Auswertung
Der Durchmesser der Hemmzonen wird auf 0,1 mm genau gemessen. Für jede Konzentration werden die Mittelwerte auf Semilogarithmenpapier aufgetragen, wobei der Logarithmus der Konzentrationen gegen den Durchmesser der Hemmzonen eingetragen wird. Für die Standardlösung sowie für den Extrakt werden die geeignetsten Geraden - wie beispielsweise nachstehend berechnet - gezogen.
Der geeignetste Punkt für das niedrigste Niveau der Standardlösung (SL) wird nach folgender Formel ermittelt:
(a) SL = (7s1 + 4s2 + s4 - 2s8) / 10
Der geeignetste Punkt für das höchste Niveau der Standardlösung (SH) wird nach folgender Formel ermittelt:
(b) SH = (7s8 + 4s4 + s2 - 2s1) / 10
Genauso werden auch die geeignetsten Punkte für das niedrigste (UL) und höchste Niveau (UH) des Extrakts ermittelt, indem in der obigen Formel - anstelle von s1, s2, s4 und s8 - u1, u2, u4 und u8 eingesetzt werden.
Die ermittelten SL- und SH-Werte werden auf dasselbe Diagramm aufgetragen. Indem man die beiden Punkte miteinander verbindet, erhält man die geeignetste Gerade für die Standardlösung. Ebenso wird mit den UL- und UH-Werten verfahren, um die geeignetste Gerade für den Extrakt zu erhalten.
In Abwesenheit von Störfaktoren sollten die Geraden parallel sein. Die Geraden können als parallel angesehen werden, wenn die Werte (SH-SL) und (UH-UL) nicht mehr als 10 v. H. vom Mittelwert abweichen.
Falls die Geraden nicht parallel sind, kann man entweder u1 und s1 oder u8 und s8 ausschalten. Dann sind die Werte, SL, SH, UL und UH mit folgenden Formeln zu ermitteln, um die geeignetste Gerade zu erhalten:
(a") SL = (5s1 +2s2 -s4) / 6 oder (5s2 +2s4 -s8) / 6
(b") SH = (5s4 + 2s2 -s1) / 6 oder (5s8 +2s4 - s2) / 6
Ebenso wird für UL und UH verfahren. Hier muss denselben Parallelitätsanforderungen entsprochen werden. Falls die Ergebnisse aus drei Niveaus ermittelt wurden, sollte dies im Analyseprotokoll vermerkt werden.
Wenn die Geraden als parallel anzusehen sind wird der Logarithmus der relativen Aktivität (log. A) durch eine der folgenden Formeln ermittelt, je nachdem, ob 3 oder 4 Niveaus für die Beurteilung der Parallelität verwendet wurden.
Für 4 Niveaus
(c)
Für 3 Niveaus
(d)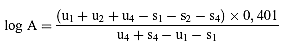
oder
(d")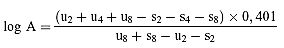
Aktivität des Probenextrakts = Aktivität der jeweiligen Standardlösung × A:
(U8 = S8 × A)
Wenn die relative Aktivität außerhalb des Bereichs der Werte 0,5 bis 2,0 liegt, wird die Bestimmung bei entsprechender Anpassung der Extraktkonzentrationen, bzw. der Standardlösungen wiederholt. Falls die relative Aktivität nicht in den Bereich der erforderlichen Werte gebracht werden kann, ist das Ergebnis als annähernd anzusehen. Dies sollte im Analyseprotokoll vermerkt werden.
Wenn die Geraden nicht als parallel anzusehen sind, wird die Bestimmung wiederholt. Wenn noch keine Parallelität erreicht wurde, ist die Bestimmung als unbefriedigend anzusehen.
Die Ergebnisse werden in Milligramm Virginiamycin je kg Futtermittel ausgedrückt.
9. Wiederholbarkeit
Der Unterschied zwischen den Ergebnissen zweier Bestimmungen eines Analytikers sollte bei ein- und derselben Probe bei Gehalten von
nicht überschreiten.
______________
1) ABl. Nr. L 170 vom 03.08.1970 S. 2.
2) ABl. Nr. L 155 vom 12.07.1971 S. 13.
3) ABl. Nr. L 279 vom 20.12.1971 S. 7.
4) Der Gehalt an Stickstoff wird nach der Halb-Mikrokjeldahlmethode bestimmt (theoretischer Gehalt: 17,7 % N).
5) Jeder handelsübliche Nährboden entsprechender Zusammensetzung, der dieselben Ergebnisse erbringt, kann verwendet werden.
6) 1 mg Virginiamycin entspricht 1 000 "UK"-Einheiten.
7) Andere Methoden, die entsprechende Bakteriensuspensionen ergeben, können angewendet werden.
8) Jeder handelsübliche Nährboden entsprechend der Zusammensetzung, der dieselben Ergebnisse erbringt, kann verwendet werden.
 |
ENDE | |
(Stand: 21.03.2024)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: ab 105.- € netto
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion