 Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 1970 -75, Lebensmittel - Futtermittel

| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 1970 -75, Lebensmittel - Futtermittel |
|
Zweite Richtlinie 71/393/EWG der Kommission vom 18. November 1971 zur Festlegung gemeinschaftlicher Analysemethoden für die amtliche Untersuchung von Futtermitteln
(ABl. Nr. L 279 vom 20.12.1971 S. 7;
RL 73/47/EWG - ABl. Nr. L 83 vom 30.03.1973 S. 35;
RL 81/680/EWG - ABl. Nr. L 246 29.08.1981 S. 32;
RL 84/4/EWG - ABl. Nr. L 15 18.01.1984 S. 28;
RL 98/64/EG - ABl. Nr. L 257 vom 19.09.1998 S. 14;
VO(EG) Nr. 152/2009 - ABl. Nr. L 54 vom::26.02.2009 S. 1aufgehoben)
aufgehoben/ersetzt gem. Art. 6 der VO (EG) 152/2009 - Entsprechungstabelle
Die Kommission der Europäischen Gemeinschaften -
gestützt auf den Vertrag zur Gründung der Europäischen Wirtschaftsgemeinschaft,
gestützt auf die Richtlinie des Rates vom 20. Juli 1970 über die Einführung gemeinschaftlicher Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln 1, insbesondere auf Artikel 2,
in Erwägung nachstehender Gründe:
Die oben genannte Richtlinie bestimmt, dass die amtlichen Untersuchungen von Futtermitteln zur Feststellung, ob die auf Grund der Rechts- und Verwaltungsvorschriften festgelegten Anforderungen hinsichtlich Beschaffenheit und Zusammensetzung der Futtermittel erfüllt sind, nach gemeinschaftlichen Probenahmeverfahren und Analysemethodendurchgeführt werden.
Die Richtlinie Nr. 71/250/EWG der Kommission vom 15. Juni 1971 2 hat bereits eine Reihe von Analysemethoden festgelegt. Nach dem Stand der seitdem durchgeführten Arbeiten ist es nunmehr geboten, eine zweite Serie von Analysemethoden festzulegen.
Die in dieser Richtlinie vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Futtermittelausschusses
- hat folgende Richtlinie erlassen:
Die Mitgliedstaaten schreiben vor, dass die Analysen für die amtliche Untersuchung von Futtermitteln auf ihren Gehalt an Feuchtigkeit, stickstoffhaltigen basen, Gesamtphosphor und Rohfett nach den in der Anlage zu dieser Richtlinie beschriebenen Methoden durchgeführt werden.
Die Mitgliedstaaten setzen spätestens zum 1. Januar 1973 die erforderlichen Rechts- und Verwaltungsvorschriften in Kraft, um den Bestimmungen dieser Richtlinie nachzukommen. Sie setzen die Kommission unverzüglich hiervon in Kenntnis.
Diese Richtlinie ist an alle Mitgliedstaaten gerichtet.
| Anlage |
1. Bestimmung von Feuchtigkeit
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Feuchtigkeitsgehalts von Futtermitteln. Sie bezieht sich nicht auf die Untersuchung von Milcherzeugnissen als Einzelfuttermittel, von Mineralstoffen und von Mischungen, die überwiegend aus Mineralstoffen bestehen, von tierischen und pflanzlichen Fetten und Ölen sowie von Ölsaaten und Ölfrüchten im Sinne der Verordnung Nr. 136/66/EWG des Rates vom 22. September 1966 über die Errichtung einer gemeinsamen Marktorganisation für Fette 3
Die Methode zur Bestimmung des Feuchtigkeitsgehalts der Ölsaaten und -früchte wird in der Anlage III der Verordnung (EWG) Nr. 1470/68 der Kommission vom 23. September 1968 über die Entnahme und Verkleinerung von Proben sowie über die Bestimmung des Gehalts der Ölsaaten an Öl, Fremdbestandteilen und Feuchtigkeit 4 festgelegt.
Die Probe wird unter definierten Bedingungen getrocknet, die von der Art des Futtermittels abhängen. Der Gewichtsverlust wird ermittelt. Bei festen Futtermitteln mit einem hohen Feuchtigkeitsgehalt ist eine zusätzliche Vortrocknung erforderlich.
3.1 Zerkleinerungsgerät aus einem Material, das keine Feuchtigkeit absorbiert, leicht zu reinigen ist, eine schnelle und gleichmäßige Zerkleinerung ermöglicht, ohne eine merkbare Erwärmung hervorzurufen, so weit wie möglich den Kontakt mit der Außenluft verhindert und den unter 4.1.1 und 4.1.2 gestellten Forderungen entspricht (z.B. Mikroschlagkreuzmühlen, Mikrozerkleinerer mit Wasserkühlung, zerlegbare Kegelmühlen, langsam laufende Kegelmühlen und Zahnscheibenmühlen);
3.2. Analysenwaage mit 0,5 mg Ablesegenauigkeit;
3.3. getrocknete Gefäße aus korrosionsbeständigem Metall oder Glas, mit luftdicht schließenden Deckeln; die Nutzfläche muss eine solche Verteilung der Probe ermöglichen, dass etwa 0,3 g auf 1 cm2 kommen;
3.4. elektrisch beheizter, temperaturgeregelter Trockenschrank (± 1 °C), der eine schnelle Regelung der Temperatur gewährleistet und eine gute Lüftung besitzt 5;
3.5. elektrisch beheizter regulierbarer Vakuumtrockenschrank mit einer Ölpumpe, der entweder mit einer Vorrichtung für die Zufuhr warmer und getrockneter Luft oder mit einem Trocknungsmittel (z.B. Calciumoxid) versehen ist;
3.6. Exsikkator mit dicker, perforierter Platte aus Metall oder aus Porzellan, der ein wirksames Trocknungsmittel enthält.
4. Verfahren
NB: Die Verfahren, die in diesem Abschnitt beschrieben werden, müssen sofort nach der Öffnung der Packungen, die die Proben enthalten, durchgeführt werden.
Die Analysen sind mindestens doppelt auszuführen.
Die Ventilation soll so beschaffen sein, dass wenn alle Weichweizenproben, die der Schrank enthalten kann, zwei Stunden lang gleichzeitig getrocknet werden, die Ergebnisse mit Bezug auf die nach 4stündiger Trocknung erzielten Ergebnisse eine unter 0,15 v. H. liegende Differenz aufweisen.
4.1. Vorbereitung
4.1.1. Futtermittel, außer den unter 4.1.2 und 4.1.3 genannten
Mindestens 50 g der Probe werden entnommen und, wenn nötig, in erforderlicher Weise unter Vermeidung von Feuchtigkeitsänderungen zerkleinert (siehe 6).
4.1.2. Getreide und Grütze
Mindestens 50 g der Probe werden entnommen. Sie wird so gemahlen, dass die Teilchengröße beim Sieben mit Maschen von 0,5 mm eine Durchlässigkeit von zumindest 50 v. H. gestattet und dass beim Sieben mit Rundmaschen von 1 mm höchstens 10 v. H. Rückstand verbleibt.
4.1.3. Flüssige oder breiige Futtermittel; Futtermittel, die überwiegend aus Fett bestehen
Etwa 25 g der Probe, auf 10 mg genau gewogen, werden entnommen, mit einer entsprechenden, auf 10 mg genau gewogenen Menge wasserfreiem Sand vermischt bis ein homogenes Produkt entsteht.
4.2. Trocknung
4.2.1. Futtermittel, außer den unter 4.2.2 und 4.2.3 genannten
Ein Gefäß mit Deckel ( 3.3) wird auf 0,5 mg genau gewogen. Etwa 5 g der Probe werden auf 1 mg genau in das tarierte Gefäß eingewogen und gleichmäßig verteilt. Das Gefäß wird nach Abnahme des Deckels in einen auf 103 °C erhitzten Trockenschrank gestellt. Damit die Temperatur des Trockenschranks nicht zu stark abfällt, ist das Gefäß möglichst rasch hineinzustellen. Es wird 4 Stunden lang getrocknet, wobei die Trocknungszeit von dem Zeitpunkt an gerechnet wird, an dem der Trockenschrank die Temperatur von 103 °C wieder erreicht hat. Nach Öffnen des Trockenschranks wird das Gefäß sofort mit dem Deckel verschlossen, aus dem Schrank genommen, zum Abkühlen 30 bis 45 Minuten lang in den Exsikkator ( 3.6) gestellt und anschließend auf 1 mg genau gewogen.
Bei Proben, die überwiegend aus Fett bestehen, wird eine zusätzliche Trocknung von 30 Minuten im Trockenschrank bei 103 °C vorgenommen. Der Unterschied zwischen den beiden Wägeergebnissen darf nicht mehr als 0,1 v. H. Feuchtigkeit betragen.
4.2.2. Getreide, Mehl, Grütze und Grieß
Ein Gefäß ( 3.3) mit Deckel wird auf 0,5 mg genau gewogen. Etwa 5 g der zerkleinerten Probe werden auf 1 mg genau in das tarierte Gefäß eingewogen und gleichmäßig verteilt. Das Gefäß wird nach Abnahme des Deckels in einen auf 130 °C erhitzten Trockenschrank gestellt. Damit die Temperatur des Trockenschranks nicht zu stark abfällt, ist das Gefäß möglichst rasch hineinzustellen. Es wird 2 Stunden lang getrocknet, wobei die Trocknungszeit von dem Zeitpunkt an gerechnet wird, an dem der Trockenschrank die Temperatur von 130 °C wieder erreicht hat. Nach öffnen des Trockenschranks wird das Gefäß sofort mit dem Deckel verschlossen, aus dem Schrank genommen, zum Abkühlen 30 bis 45 Minuten lang in den Exsikkator ( 3.6) gestellt und anschließend auf 1 mg genau gewogen.
4.2.3. Mischfuttermittel mit einem Anteil von mehr als 4 v. H. Saccharose oder Laktose sowie folgende Einzelfuttermittel: Johannisbrotschrot, hydrolysierte Getreideerzeugnisse, Malzkeime, Zuckerrübenschnitzel, Fischpresssaft, Zucker und Mischfuttermittel von mehr als 25 v. H. kristallwasserhaltigen Mineralstoffen
Ein Gefäß ( 3.3) mit Deckel wird auf 0,5 mg genau gewogen. Etwa 5 g der Probe werden auf 1 mg genau in das tarierte Gefäß eingewogen und gleichmäßig verteilt. Das Gefäß wird nach Abnahme des Deckels in einen auf 80 bis 85 °C erhitzten Vakuumtrockenschrank ( 3.5) gestellt. Damit die Temperatur des Vakuumtrockenschranks nicht zu stark abfällt, ist das Gefäß möglichst rasch hineinzustellen.
Der Druck wird auf 100 Torr eingestellt und die Probe 4 Stunden lang bei diesem Druck entweder unter Zufuhr von heißer trockener Luft oder mittels eines Trocknungsmittels (etwa 300 g für 20 Proben) getrocknet. Im letzten Fall wird beim Erreichen des vorgeschriebenen Drucks die Verbindung zur Vakuumpumpe unterbrochen. Die Trocknungszeit wird von dem Zeitpunkt an gerechnet, an dem der Trockenschrank die Temperatur von 80 ° bis 85 °C wieder erreicht hat. Nach Ablauf der Trocknungszeit wird der Vakuumtrockenschrank vorsichtig auf athmosphärischen Druck gebracht. Nach öffnen des Vakuumtrockenschranks wird das Gefäß sofort mit dem Deckel verschlossen, aus dem Schrank genommen, zum Abkühlen 30 bis 45 Minuten lang in den Exsikkator ( 3.6) gestellt und anschließend auf 1 mg genau gewogen. Es wird weitere 30 Minuten unter Vakuum im Trockenschrank bei 80 ° bis 85 °C getrocknet und erneut gewogen. Der Unterschied zwischen den beiden Wägeergebnissen darf nicht mehr als 0,1 v. H. Feuchtigkeit betragen.
4.3. Vortrocknung
4.3.1. Futtermittel, außer den unter 4.3.2 genannten
Eine Vortrocknung ist bei festen Futtermitteln mit einem hohen Feuchtigkeitsgehalt, deren Zerkleinerung schwierig ist, wie folgt vorzunehmen:
50 g, auf 10 mg genau, der ungemahlenen Probe (eine grobe Zerkleinerung wird, wenn nötig, bei gepressten oder brockenhaltigen Futtermitteln vorgenommen), werden in einen geeigneten Behälter (z.B. eine Aluminiumschale 20 × 12 cm mit einem Rand von 0,5 cm) eingewogen. In einem Trockenschrank wird bei einer Temperatur von 60 ° bis 70 °C getrocknet, bis der Feuchtigkeitsgehalt einen Wert zwischen 8 und 12 v. H. erreicht hat. Der Behälter wird aus dem Trockenschrank herausgenommen, man lässt 1 Stunde lang offen abkühlen, anschließend wird auf 10 mg genau gewogen. Je nach der Art des Futtermittels wird, wie bei 4.1.1 beschrieben, sofort danach zerkleinert und, wie bei 4.2.1 oder 4.2.3 beschrieben, getrocknet.
4.3.2. Getreide
Körner, deren Feuchtigkeitsgehalt höher als 17 v. H. ist, müssen wie folgt vorgetrocknet werden:
50 g der ungemahlenen Körner werden auf 10 mg genau in einen geeigneten Behälter (z.B. eine Aluminiumschale 20 × 12 cm mit einem Rand von 0,5 cm) eingewogen, in einem Trockenschrank 5 bis 7 Minuten lang bei einer Temperatur von 130 °C getrocknet und aus dem Trockenschrank herausgenommen. Man lässt im Labor 2 Stunden lang offen abkühlen und wiegt dann auf 10 mg genau. Sofort danach wird nach 4.1.2 gemahlen und, wie bei 4.2.2 beschrieben, getrocknet.
5. Berechnung der Ergebnisse
Der Feuchtigkeitsgehalt der Probe inv. H. wird durch die folgenden Formeln gegeben:
5.1. Trocknung ohne Vortrocknung
(E - m ) * 100 / E
E = Anfangsmasse der Probe in Gramm,
m = Masse der trockenen Probe in Gramm.
5.2. Trocknung mit Vortrocknung
[(M" - m ) M] / M" + E - M * 100 / E = 100 1 - Mm / EM"
E = Anfangsmasse der Probe in Gramm,
M = Masse der Probe in Gramm nach der Vortrocknung,
M" = Masse der Probe in Gramm nach der Zerkleinerung und dem Zermahlen,
m = Masse der trockenen Probe in Gramm.
5.3. Wiederholbarkeit
Der Unterschied zwischen zwei Parallelbestimmungen darf bei ein und derselben Probe 0,2 v. H. Feuchtigkeit nicht überschreiten.
6. Bemerkung
Wenn eine Zerkleinerung sich als notwendig erweist und dabei mit einer Änderung des Feuchtigkeitsgehalts des Materials gerechnet werden muss, so sind die Analysenergebnisse, welche die Bestandteile des Futtermittels betreffen, auf den Feuchtigkeitsgehalt der Originalprobe umzurechnen.
2. Bestimmung von flüchtigen stickstoffhaltigen basen
A. Bestimmung durch Mikrodiffusion
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an flüchtigen stickstoffhaltigen basen, ausgedrückt als Ammoniak, in Futtermitteln.
2. Prinzip
Die Probe wird mit Wasser extrahiert, die Lösung geklärt und filtriert. Die flüchtigen stickstoffhaltigen basen werden nach Zusatz von Kaliumcarbonatlösung durch Mikrodiffusion abgetrennt, in einer Borsäurelösung aufgefangen und mit Schwefelsäure titriert.
3. Reagenzien
3.1. Trichloressigsäurelösung 20 v. H. (G/V);
3.2. Indikator: 33 mg Bromkresolgrün und 65 mg Methylrot werden in 100 ml Äthanol 95 bis 96 v. H. (V/V) gelöst;
3.3. Borsäurelösung: 10 g Borsäure p.a. werden in einem 1-l-Meßkolben in 200 ml Äthanol 95 bis 96 v. H. (V/V) und 700 ml Wasser gelöst. Vom Indikator ( 3.2) werden 10 ml hinzugefügt. Die Lösung wird gemischt und nötigenfalls unter Zusatz von Natriumhydroxidlösung auf eine hell-rote Farbe gebracht. 1 ml dieser Lösung kann bis zu 300 µg NH3 binden;
3.4. Gesättigte Kaliumkarbonatlösung: 100 g Kaliumcarbonat p.a. werden in 100 ml siedendem Wasser gelöst; nach dem Abkühlen wird die Lösung filtriert;
3.5. 0,02 N Schwefelsäure.
4. Geräte
4.1. Mechanischer Schüttelapparat: ungefähr 35 bis 40 U/Min,
4.2. Conwayschalen (vgl. Skizze) aus Glas oder Plastik,
4.3. Mikrobüretten 1/100 ml Ablesegenauigkeit.
5. Ausführung
10 g der Probe werden auf 1 mg genau gewogen, mit 100 ml Wasser in einen 200-ml-Meßkolben gegeben und 30 Minuten lang im Schüttelapparat geschüttelt. Dann werden 50 ml Trichloressigsäurelösung ( 3.1) hinzugefügt, mit Wasser bis zur Marke aufgefüllt, kräftig geschüttelt und durch ein Faltenfilter filtriert.
In die Mitte der Conwayschale wird 1 ml Borsäurelösung ( 3.3) pipettiert, in den Ring der Schale 1 ml des Probenfiltrats. Die Schale wird mit dem angefetteteten Deckel teilweise bedeckt, dann gibt man schnell 1 ml der gesättigten Kaliumcarbonatlösung ( 3.4) in den Ring und verschließt die Schale luftdicht. Um mit Sicherheit eine Mischung der beiden Reagenzien zu erreichen, wird die Schale vorsichtig in waagerechter Stellung gedreht. Darauf lässt man das Ganze mindestens 4 Stunden lang bei Raumtemperatur oder 1 Stunde lang bei 40 °C stehen.
Die in der Borsäurelösung enthaltenen flüchtigen basen werden anschließend mit 0,02 N Schwefelsäure ( 3.5) unter Verwendung einer Mikrobürette ( 4.3) titriert.
Nach demselben Verfahren ist ein Blindversuch, ohne die zu analysierende Probe, durchzuführen.
6. Berechnung der Ergebnisse
1 ml 0,02 Schwefelsäure entspricht 0,34 mg Ammoniak. Das Ergebnis wird in Hundertteilen der Probe ausgedrückt. Wiederholbarkeit
Der Unterschied zwischen zwei Parallelbestimmungen darf bei ein und derselben Probe bei Gehalten von weniger als 1,0 v. H. Ammoniak 10 v. H. relativ, bei Gehalten von 1,0 v. H. und mehr 0,1 v. H. absolut nicht überschreiten.
7. Bemerkung
Bei einem Gehalt der Probe von mehr als 0,6 v. H. Ammoniak ist das ursprüngliche Filtrat zu verdünnen
. CONWAY-SCHALE
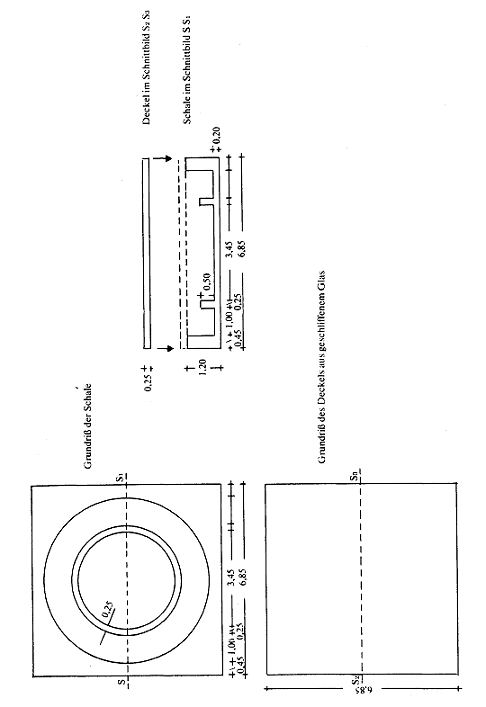
B. Bestimmung durch Destillation
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an flüchtigen stickstoffhaltigen basen, ausgedrückt als Ammoniak, in Fischmehl, das praktisch keinen Harnstoff enthält. Sie ist nur anzuwenden bei Gehalten kleiner als 0,25 v. H. Ammoniak.
2. Prinzip
Die Probe wird mit Wasser extrahiert, die Lösung geklärt und filtriert. Die flüchtigen stickstoffhaltigen basen werden nach Zusatz von Magnesiumoxid in der Siedehitze abgetrennt und in einer definierten Menge Schwefelsäure aufgefangen, deren Überschuß durch Natriumhydroxidlösung zurücktitriert wird.
3. Reagenzien
3.1. Trichloressigsäurelösung 20 v. H. (G/V),
3.2. Magnesiumoxid p.a.,
3.3. Antischaumemulsion (z.B. Silikon),
3.4. 0,1 N Schwefelsäure,
3.5. 0,1 N Natriumhydroxidlösung,
3.6. Methylrotlösung 0,3 v. H. (G/V) in Äthanol 95 - 96 v. H. (V/V).
4. Geräte
4.1. Mechanischer Schüttelapparat, ungefähr 35 bis 40 U/Min.
4.2. Destillationsapparatur nach Kjeldahl.
5. Ausführung
10 g der Probe werden, auf 1 mg genau gewogen, mit 100 ml Wasser in einen 200-ml-Meßkolben gegeben und 30 Minuten lang im Schüttelapparat geschüttelt.
Dann werden 50 ml Trichloressigsäurelösung ( 3.1) hinzugefügt, mit Wasser bis zur Marke aufgefüllt, kräftig geschüttelt und durch ein Faltenfilter filtriert.
Je nach dem vermuteten Gehalt an flüchtigen stickstoffhaltigen basen wird eine entsprechende Menge (im allgemeinen 100 ml) des klaren Filtrats auf 200 ml verdünnt und 2 g Magnesiumoxid ( 3.2) sowie einige Tropfen Antischaumemulsion ( 3.3) hinzugefügt. Die Lösung muss gegen Lackmuspapier alkalisch reagieren; anderenfalls ist noch Magnesiumoxid ( 3.2) zuzusetzen. In der Kjeldahl-Apparatur werden etwa 150 ml der Lösung abdestilliert und das Destillat in einem Erlenmeyerkolben aufgefangen, der eine genau definierte Menge (zwischen 25 und 50 ml) 0,1 N Schwefelsäure ( 3.4) enthält. Bei der Destillation ist eine Überhitzung der Kolbenwände zu vermeiden. Die schwefelsaure Lösung wird 2 Minuten lang aufgekocht, dann abgekühlt; der Überschuss an Schwefelsäure wird mit 0,1 N Natriumhydroxidlösung ( 3.5) nach Zugabe von Methylrot ( 3.6) als Indikator zurücktitriert.
Nach demselben Verfahren ist ein Blindversuch, ohne die zu analysierende Probe, durchzuführen.
6. Berechnung der Ergebnisse
1 ml 0,1 Schwefelsäure entspricht 1,7 mg Ammoniak.
Das Ergebnis wird in Hundertteilen der Probe ausgedrückt. Wiederholbarkeit
Der Unterschied zwischen zwei Parallelbestimmungen darf bei ein und derselben Probe 10 v. H. (relativ) Ammoniak nicht überschreiten.
3. Bestimmung von Gesamtphosphor
- photometrische Methode -
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an Gesamtphosphor in Futtermitteln. Sie ist besonders für die Untersuchung von Futtermitteln mit einem geringen Gehalt an Phosphor geeignet. In bestimmten Fällen (phosphorreiche Erzeugnisse) kann eine gravimetrische Methode angewandt werden.
2. Prinzip
Die Probe wird entweder auf trockenem Wege (besonders bei organischen Futtermitteln) oder auf nassem Wege (besonders bei Mineralstoffen und flüssigen Futtermitteln) aufgeschlossen und in Säure gelöst. Die Lösung wird mit dem Vanadatmolybdat-Reagenz behandelt. Die Extinktion der gelb gefärbten Lösung wird bei 430 nm im Spektralphotometer gemessen.
3. Reagenzien
3.1. Calciumcarbonat p. a.,
3.2. Salzsäure p. a., D: 1,1 (etwa 6 N),
3.3. Salpetersäure p. a., D: 1,045,
3.4. Salpetersäure p. a., D: 1,38 bis 1,42,
3.5. Schwefelsäure p. a., D: 1,84.
3.6. Vanadatmolybdat-Reagenz: 200 ml Ammoniumheptamolybdatlösung ( 3.6.1) werden mit 200 ml Ammoniummonovanadatlösung ( 3.6.2) und 134 ml Salpetersäure ( 3.4) in einem 1-l-Meßkolben gemischt und bis zur Marke mit Wasser aufgefüllt.
3.6.1. Ammoniumheptamolybdatlösung: 100 g Ammoniumheptamolybdat p. a., (NH4)6Mo7O24 · 4H2O, werdeninwarmem Wasser gelöst. 10 ml Ammoniak (D: 0,91) werden hinzugefügt, und das Volumenwird mit Wasser auf 1 l ergänzt.
3.6.2. Ammoniummonovanadatlösung: 2,35 g Ammoniummonovanadat p. a, NH4VO3, werden in 400 ml warmem Wasser gelöst. 20 ml verdünnte Salpetersäure (7 ml HNO3 ( 3.4) + 13 ml H2O) werden unter Rühren langsam hinzugefügt, und das Volumen wird auf 1 l mit Wasser ergänzt.
3.7. Phosphor-Standardlösung 1 mg pro ml: 4,387 g Kaliumdihydrogenphosphat p. a., KH2PO4, werden in Wasser gelöst und auf 1 l mit Wasser aufgefüllt.
4. Geräte
4.1. Veraschungsschalen aus Quarz oder Porzellan,
4.2. elektrischer Muffelofen mit Thermostat auf 550°C geregelt,
4.3. Kjeldahlkolben, 250 ml,
4.4. Messkolben und Präzisions-Pipette,
4.5. Spektralphotometer,
4.6. Reagenzgläser, ca. 16 mm Durchmesser mit N 14,5; Fassungsvermögen: 25 - 30 ml.
5. Ausführung
5.1. Vorbereitung der Lösung
Je nach der Art der Probe wird die Lösung, wie unter 5.1.1 oder 5.1.2 angegeben, vorbereitet.
5.1.1. Allgemeines Verfahren
1 g oder mehr der Probe werden auf 1 mg genau eingewogen und in einen Kjeldahlkolben gebracht. 20 ml Schwefelsäure ( 3.5) werden hinzugefügt; dann wird umgeschüttelt, um das Material mit der Säure gut zu benetzen und zu verhindern, dass es an den Wandungen des Kolbens anhaftet; anschließend wird erhitzt und 10 Minuten im Sieden gehalten. Nach geringem Abkühlen werden 2 ml Salpetersäure ( 3.4) hinzugefügt; es wird schwach erhitzt und wieder etwas abgekühlt. Dann wird erneut wenig Salpetersäure ( 3.4) hinzugegeben, zum Sieden erhitzt und das Verfahren so oft wiederholt, bis die Lösung farblos geworden ist. Hierauf wird abgekühlt, etwas Wasser zugesetzt und die Flüssigkeit in einen 500-ml-Meßkolben überführt, wobei der Kjeldahlkolben mit warmem Wasser ausgespült wird. Nach Abkühlen wird zur Marke aufgefüllt, umgeschüttelt und filtriert.
5.1.2. Verfahren für Proben, die organische Stoffe enthalten, aber frei sind von Calcium- bzw. Magnesium-dihydrogenphosphaten
Etwa 2,5 g der Probe werden auf 1 mg genau in eine Veraschungsschale eingewogen und mit 1 g Calciumcarbonat ( 3.1) innig gemischt. Im Muffelofen wird bei 550°C ± 5°C verascht, bis die Asche weiß oder grau ist (kleine Mengen Kohle stören nicht).
Die Asche wird in ein250-ml-Becherglas gebracht, 20 ml Wasser und soviel Salzsäure ( 3.2) zugefügt, bis das Aufbrausen ausbleibt; zuletzt wird ein Überschuss von10 ml Salzsäure ( 3.2) zugegeben. Dann wird das Becherglas auf ein Sandbad gestellt und bis zur Trockne eingedampft, um die Kieselsäure unlöslich zu machen. Der Rückstand wird in 10 ml Salpetersäure ( 3.3) aufgenommen und 5 Minuten lang auf dem Sandbad zum Sieden erhitzt, wobei der Rückstand nicht ganz trocken werden soll. Die Flüssigkeit wird in einen 500-ml-Meßkolben übergespült, wobei das Becherglas mehrmals mit heißem Wasser ausgewaschen wird. Nach Abkühlen wird mit Wasser zur Marke aufgefüllt, umgeschüttelt und filtriert.
5.2. Entwicklung der Färbung und Messung der Extinktion
Ein aliquoter Teil des nach 5.1.1 oder 5.1.2 erhaltenen Filtrats wird verdünnt, um eine Konzentration an Phosphor von höchstens 40 µg/ml zu erhalten. 10 ml dieser Lösung werden in ein Reagenzglas mit Schliff ( 4.6) pipettiert und 10 ml Vanadatmolybdat-Reagenz ( 3.6) hinzugefügt. Man schüttelt um und lässt mindestens 10 Minuten lang bei einer Temperatur von 20°C stehen. Anschließend wird die Extinktion im Spektralphotometer bei 430 nm gegen eine Lösung von 10 ml Wasser und 10 ml Vanadatmolybdat-Reagenz ( 3.6) gemessen.
5.3. Eichkurve
Aus der Standardlösung ( 3.7) werden Lösungen, die 5, 10, 20, 30 und 40 µg Phosphor pro ml enthalten, hergestellt. Zu je 10 ml dieser Lösungen werden 10 ml Vanadatmolybdat-Reagenz (3.6) hinzugefügt. Man schüttelt um und lässt mindestens 10 Minuten lang bei einer Temperatur von 20°C stehen. Dann wird die Extinktion unter den bei 5.2 angegebenen Bedingungen gemessen. Die Eichkurve wird aufgestellt, indem die Extinktionswerte in die Ordinate und die entsprechende Phosphormenge in die Abszisse eingetragen werden. Die Kurve ist linear für die Konzentrationen an Phosphor von 0 bis 40 µg/ ml.
6. Berechnung der Ergebnisse
Der Gehalt an Phosphor der Analysenprobe wird unter Benutzung der Eichkurve bestimmt.
Das Ergebnis wird in Hundertteilen der Probe ausgedrückt.
Wiederholbarkeit
Der Unterschied zwischen zwei Parallelbestimmungen darf bei ein und derselben Probe bei Gehalten von
weniger als 5 v. H. Phosphor 3 v. H. relativ, bei Gehalten
von 5 v. H. Phosphor und mehr 0,15 v. H. absolut nicht überschreiten.
4. Bestimmung von Rohfett
1. Zweck und Anwendungsbereich
Die Methode dient zur Bestimmung des Rohfettgehalts von Futtermitteln. Sie bezieht sich nicht auf die Untersuchung von Ölsaaten und Ölfrüchten im Sinne der Verordnung (EWG) Nr. 136/66 des Rates.
Je nach Art und Zusammensetzung des Futtermittels sowie dem Zweck der Untersuchung ist eines der beiden nachstehend beschriebenen Verfahren anzuwenden.
1.1. Verfahren a - Direkt extrahierbares Rohfett
Das Verfahren ist anzuwenden bei Einzelfuttermitteln pflanzlicher Herkunft mit Ausnahme derjenigen, die in den Anwendungsbereich von Verfahren B fallen.
1.2. Verfahren B - Gesamtrohfett
Das Verfahren ist anzuwenden bei Einzelfuttermitteln tierischen Ursprungs und bei allen Mischfuttermitteln. Es ist außerdem bei allen Materialien anzuwenden, aus denen Fett ohne vorherige Hydrolyse nicht vollständig extrahiert werden kann, z.B. Kleber, Hefen, Kartoffeleiweiß und Erzeugnissen, die Verfahren unterzogen wurden wie Extrudieren, Flockieren und Erhitzen.
1.3. Auswertung
In allen Fällen, in denen mit Verfahren B ein höheres Ergebnis erzielt wird als mit Verfahren A, gilt das mit Verfahren B erhaltene Ergebnis als der richtige Wert.
2. Prinzip
2.1. Verfahren A
Die Probe wird mit Petrolether extrahiert. Das Lösungsmittel wird abdestilliert und der Rückstand getrocknet und gewogen.
2.2. Verfahren B
Die Probe wird heiß mit Salzsäure behandelt. Die Mischung wird abgekühlt und filtriert. Der gewaschene und getrocknete Rückstand wird nach dem Verfahren a weiterbehandelt.
3. Reagenzien
3.1. Petrolether mit einem Siedebereich von 40-60 °C. Die Bromzahl muss weniger als 1 und der Abdampfrückstand weniger als 2 mg/100 ml betragen.
3.2. Natriumsulfat, wasserfrei.
3.3. Salzsäure, c = 3 mol HCl/l.
3.4. Filtrationsmittel, z.B. Kieselgur, Hyflosupercel.
4. Geräte
4.1. Extraktionsapparat: Sofern das Gerät mit einem Siphon (Soxhletapparat) ausgestattet ist, muss die Rückflussmenge so bemessen sein, dass das Lösungsmittel mindestens zehnmal in der Stunde abläuft; wenn es sich um ein Gerät ohne Siphon handelt, muss die Rückflussmenge etwa 10 ml pro Minute betragen.
4.2. Extraktionshülsen: Diese müssen frei sein von petroletherlöslichen Stoffen und eine Porosität besitzen, die den Forderungen unter 4.1 entspricht.
4.3. Trockenschrank: Vakuumtrockenschrank, einstellbar auf 75 °C ± 3 °C, oder Lufttrockenschrank, einstellbar auf 100 °C ± 3 °C.
4.4. Mikrowellengerät.
5. Ausführung
5.1. Verfahren a (für Proben mit hohem Fettgehalt siehe Bemerkung 8.1)
5 g der Probe werden auf 1 mg genau abgewogen, dann in eine Extraktionshülse ( 4.2) übergeführt und mit einem fettfreien Wattebausch abgedeckt.
Die Hülse wird in einen Extraktionsapparat ( 4.1) gebracht und sechs Stunden mit Petrolether ( 3.1). extrahiert, wobei der Petroletherextrakt in einem trockenen, mit einigen Stückchen Bimsstein 6 versehenen, tarierten Kolben aufgefangen wird.
Nach beendeter Extraktion wird das Lösungsmittel abdestilliert, der Rückstand mit Kolben anschließend 1,5 Stunden in einem Trockenschrank ( 4.3) getrocknet und nach dem Abkühlen in einem Exsikkator gewogen. Durch eine zweite Trocknung von 30 Minuten Dauer ist zu prüfen, ob das Gewicht des Fettes konstant geblieben ist (der Gewichtsverlust zwischen zwei aufeinander folgenden Wägungen muss unter 1 mg bleiben).
5.2. Verfahren B
2,5 g der Probe (siehe Bemerkung 8.2) werden auf 1 mg genau abgewogen und in ein 400-ml-Becherglas oder einen 300-ml-Erlenmeyerkolben gebracht. Anschließend werden 100 ml Salzsäure, c = 3 mol/l ( 3.3), und einige Stückchen Bimsstein hinzugefügt. Das Becherglas wird mit einem Uhrglas abgedeckt, bzw. dem Erlenmeyerkolben wird ein Rückflusskühler aufgesetzt. Das Gemisch wird auf kleiner Flamme oder auf einer Heizplatte bis zum Siedepunkt erhitzt und eine Stunde schwach siedengelassen. Es muss vermieden werden, dass das Material an den Wänden des Gefäßes ansetzt.
Dann wird abgekühlt und so viel vom Filtrationsmittel ( 3.4) zugesetzt, dass beim Filtrieren kein Fettverlust eintritt. Filtriert wird unter Verwendung zweier ineinander gelegter feuchter fettfreier Papierfilter. Der Rückstand wird bis zur neutralen Reaktion mit kaltem Wasser gewaschen. Danach ist zu prüfen, ob das Filtrat noch Fett enthält. Vorhandensein von Fett zeigt, dass vor der Hydrolyse eine Extraktion der Probe mit Petrolether nach Verfahren a vorzunehmen ist.
Der doppelte Papierfilter mit dem Rückstand ist auf ein Uhrglas zu legen und ca. 1,5 Stunden bei 100 °C ± 3 °C im Trockenschrank oder bei 75 °C ± 3 °C im Vakuumtrockenschrank ( 4.3) zu trocknen.
Der doppelte Filter mit dem trockenen Rückstand wird in eine Extraktionshülse ( 4.2) gebracht und mit einem fettfreien Wattebausch bedeckt. Die Hülse wird in einen Extraktionsapparat ( 4.1) eingesetzt. Dann wird wie unter 5.1 Unterabsätze 2 und 3 beschrieben fortgefahren.
6. Berechnung der Ergebnisse
Das Gewicht des Rückstands wird in Hundertteilen (Prozenten) der Probe ausgedrückt.
7. Wiederholbarkeit
Der Unterschied zwischen den Ergebnissen zweier Parallelbestimmungen (eines Analytikers) sollte bei derselben Probe bei Gehalten an Rohfett von
nicht überschreiten
8. Bemerkungen
8.1. Bei Erzeugnissen mit hohem Fettgehalt, die schwer zu zerkleinern sind oder sich zur Entnahme einer reduzierten homogenen Probe nicht eignen, ist folgendermaßen zu verfahren:
20 g der Probe werden auf 1 mg genau abgewogen und mit 10 g oder mehr wasserfreiem Natriumsulfat ( 3.2) gemischt. Dann wird mit Petrolether ( 3.1) wie unter 5.1 beschrieben extrahiert. Der dabei aufgefangene Extrakt wird mit Petrolether ( 3.1) auf 500 ml aufgefüllt und gemischt. Von der Lösung werden 50 ml entnommen und in einen kleinen, trockenen, mit Bimssteinkörnchen versehenen6 und tarierten Kolben gebracht. Das Lösungsmittel wird abdestilliert; dann wird wie unter 5.1 letzter Unterabsatz beschrieben getrocknet und weiterverfahren.
Der Extraktionsrückstand in der Hülse wird vom Lösungsmittel befreit und auf 1 mm zerkleinert. Das Material wird in die Extraktionshülse zurückgebracht (kein Natriumsulfat hinzufügen). Dann wird wie unter 5.1 Unterabsätze 2 und 3 beschrieben fortgefahren.
Der Rohfettgehalt, ausgedrückt in Hundertteilen der Probe, wird nach folgender Formel ermittelt:
(10 a + b) × 5
Darin sind:
a = Rückstandsmasse in Gramm nach der ersten Extraktion (aliquoter Teil des Extraktes),
b = Rückstandsmasse in Gramm nach der zweiten Extraktion.
8.2. Bei fettarmen Erzeugnissen kann mit einer Einwaage von 5 g gearbeitet werden.
8.3. Heimtierfutter mit hohem Wassergehalt müssen vor der Hydrolyse und Extraktion nach Verfahren B möglicherweise mit wasserfreiem Natriumsulfat gemischt werden.
8.4. In 5.2 kann die Verwendung von heißem statt kaltem Wasser zum Waschen des Rückstands nach der Filtration möglicherweise wirksamer sein.
8.5. Die Trocknungszeit von 1,5 Stunden muss bei einigen Futtermitteln möglicherweise verlängert werden. Übermäßiges Trocknen ist zu vermeiden, da dies zu niedrigen Ergebnissen führen kann. Es kann auch ein Mikrowellengerät ( 4.4) verwendet werden.
8.6. Bei einem Rohfettgehalt von mehr als 15 v. H. wird eine Extraktion nach Verfahren a vor der Hydrolyse und eine erneute Extraktion nach Verfahren B empfohlen.
______________________
1) ABl. Nr. L 170 vom 03.08.1970 S. 2.
2) ABl. Nr. L 155 vom 12.07.1971 S. 13.
3) ABl. Nr. 172 vom 30.09.1966 S. 3025/66.
4) ABl. Nr. L 239 vom 28.09.1968 S. 2.
5) Für die Trocknung von Getreide, Mehl, Grütze und Grieß soll der Trockenschrank eine solche Wärmekapazität haben, dass er, wenn er auf eine Temperatur von 131 °C eingestellt worden ist, diese Temperatur in weniger als 45 Minuten wieder erreicht, nachdem die Höchstzahl gleichzeitig zu trocknender Proben hineingestellt wurde.
6) Wenn das Fett später qualitativ untersucht werden soll, sind die Bimssteinstückchen durch Glasperlen zu ersetzen.
 |
ENDE | |
(Stand: 21.03.2024)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: ab 105.- € netto
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion