

umwelt-online: Bestimmung der Toxizität
| |
zurück | |
| Methoden zur Bestimmung der Toxizität
B.1. bis - Akute orale Toxizität - Fest-Dosis-Methode |
Anhang V zur RL 67/548/EWG |
B.1.bis 1. Methode
Diese Prüfmethode entspricht der OECD TG 420 (2(X)1).
B.1.bis 1.1 Einleitung
Herkömmliche Methoden zur Bewertung der akuten Toxizität verwenden den Tod der Versuchstiere als Endpunkt. 1984 hat die British Toxicology Society einen neuen Ansatz zur Bewertung der akuten Toxizität vorgeschlagen, der von der Verabreichung einer Serie fester Dosen ausging (1). Dieser Ansatz bezog sich nicht auf den Tod der Versuchstiere als Endpunkt, sondern stützte sich auf die Beobachtung deutlicher Toxizitätszeichen, die bei einer bestimmten Dosis aus einer Serie fester Dosierungen auftraten. Nach In-vivo-Validierungsstudien im Vereinigten Königreich (2) sowie in internationalem Rahmen (3) wurde im Jahre 1992 als Prüfmethode anerkannt. Später wurden die statistischen Eigenschaften der Fest-Dosis-Methode mit Hilfe mathematischer Modelle in einer Reihe von Studien berechnet (4)(5)(6). Mit In-vivo-Studien und anhand von Modellstudien konnte nachgewiesen werden, dass das Verfahren reproduzierbar ist, mit einer geringeren Anzahl an Versuchstieren auskommt und weniger Leiden verursacht als die herkömmlichen Methoden und eine ähnliche Bewertung der Substanzen ermöglicht wie die sonstigen Methoden zur Prüfung der akuten Toxizität.
Leitlinien für die Auswahl der geeignetsten Testmethode für einen bestimmten Zweck enthält das Guidance Document an Acute Oral Toxicity Testing (7). Dieser Leitfaden beinhaltet darüber hinaus weitere Informationen zur Durchführung und Auswertung der Prüfmethode B.1 bis.
Eines der Prinzipien der Methode besteht darin, dass in der Hauptstudie nur mäßig toxische Dosen eingesetzt werden und dass die Verabreichung von voraussichtlich letalen Dosen vermieden werden sollte. Außerdem brauchen keine Dosierungen verabreicht zu werden, die aufgrund ätzender oder stark reizender Wirkungen bekanntermaßen starke Schmerzen und schweres Leiden verursachen. Moribunde Tiere oder Tiere, die offensichtlich unter Schmerzen leiden oder Anzeichen von schwerem und anhaltendem Leiden zeigen, sind auf humane Weise zu töten und werden bei der Auswertung der Testergebnisse auf die gleiche Weise gewertet wie während des Tests gestorbene Tiere. Kriterien für die Entscheidung, moribunde oder schwer leidende Tiere zu töten, sowie Hinweise zur Erkennung des absehbaren oder bevorstehenden Todes sind Gegenstand eines gesonderten Leitfadens (8).
Die Methode führt zu Informationen über die gefährlichen Eigenschaften und ermöglicht die Bewertung und Klassifizierung der Substanz gemäß dem Globalen Harmonisierten System (GHS) zur Klassifizierung chemischer Stoffe mit akut toxischer Wirkung (9).
Das Prüflabor soll vor der Durchführung der Studie sämtliche verfügbaren Informationen berücksichtigen. Zu diesen Informationen gehören die Identität und die chemische Struktur der zu testenden Substanzen, die physikalisch-chemischen Eigenschaften der Substanzen, die Ergebnisse sonstiger In-vivo- oder In-vitro-Toxizitätstests in Verbindung mit den betreffenden Substanzen, toxikologische Daten über strukturverwandte Substanzen sowie die vorgesehene(n) Verwendung(en) der Substanzen. Diese Informationen werden benötigt, um die zuständigen Stellen zu überzeugen, dass der Test für den Schutz der menschlichen Gesundheit wichtig ist und bei der Wahl der geeigneten Startdosis hilft.
B.1.bis 1.2 Definitionen
Akute orale Toxizität: Schädliche Wirkungen, die nach der oralen Verabreichung einer Einzeldosis oder mehrerer innerhalb von 24 Stunden verabreichter Dosen einer Substanz auftreten.
Verzögerter Tod: Das betreffende Tier stirbt zwar nicht binnen 48 Stunden und wirkt in diesem Zeitraum nicht moribund; der Tod tritt jedoch später während des 14-tägigen Beobachtungszeitraums ein.
Dosis: Verabreichte Menge der Testsubstanz; die Dosis wird als Gewicht der Testsubstanz pro Gewichtseinheit des Versuchstiers ausgedrückt (z.B. mg/kg).
Evidente Toxizität: Ein allgemeiner Begriff, der deutliche Anzeichen von Toxizität nach der Verabreichung einer Testsubstanz dahingehend beschreibt, dass bei der nächsthöheren festen Dosis entweder starke Schmerzen und anhaltende Anzeichen für schweres Leiden, ein moribunder Zustand (Kriterien siehe Humane Endpoints Guidance Document (8)) oder wahrscheinlich der Tod der meisten Versuchstiere zu erwarten sind (siehe z.B. (3)).
GHS: Globally Harmonised Classification System for Chemical Substances and Mixtures (Weltweit harmonisiertes Klassifizierungssystem für chemische Substanzen und Gemische); ein gemeinsames Projekt von OECD (Menschliche Gesundheit und Umwelt), UN-Expertenausschuss für den Transport von Gefahrgütern (physikalisch-chemische Eigenschaften) und ILO (Gefahrenanzeige) koordiniert im Rahmen des Interorganisation Programme for the Sound Management of Chemicals (IOMC).
Bevorstehender Tod: Der moribunde Zustand oder Tod ist vor dem nächsten vorgesehenen Beobachtungszeitpunkt zu erwarten. Anzeichen für diesen Zustand können bei Nagetieren Krämpfe, Seitenlage, liegende Stellung und Tremor sein (siehe Humane Endpoint Guidance Document (8)).
LD50 (mittlere Letaldosis): Statistisch ermittelte Einzeldosis einer Substanz, bei der davon ausgegangen werden kann, dass sie bei oraler Verabreichung den Tod von 50 % der Tiere verursacht; der LD50-Wert wird als Gewicht der Testsubstanz pro Gewichtseinheit der Versuchstiere ausgedrückt (mg/kg).
Limit-Dosis: Dosis am oberen Grenzwert für den betreffenden Versuch (2.000 oder 5.000 mg/kg)
Moribunder Zustand: Zustand des Sterbens oder des Unvermögens, (selbst bei Behandlung) zu überleben (siehe Humane Endpoint Guidance Document (8))
Voraussagbarer Tod: Vorhandensein klinischer Zeichen, die auf den Eintritt des Todes zu einem bekannten Zeitpunkt in der Zukunft - vor dem planmäßigen Ende des Experiments - hinweisen, z.B. das Unvermögen, Wasser oder Nahrung aufzunehmen (siehe Humane Endpoint Guidance Document (8))
B.1.bis 1.3 Prinzip der Prüfmethode
An Gruppen von Versuchstieren eines Geschlechts wird in einem schrittweisen Verfahren jeweils die feste Dosis 5, 50, 300 und 2.000 mg/kg verabreicht. (Im Ausnahmefall kann eine weitere feste Dosis von 5.000 mg/kg in Betracht gezogen werden (siehe Abschnitt 1.6.2)). Als Startdosis wird auf der Grundlage einer Vorstudie die Dosis gewählt, die voraussichtlich gewisse Toxizitätsanzeichen verursachen wird, ohne schwere toxische Wirkungen oder Mortalität hervorzurufen. Klinische Anzeichen und Zustände, die mit Schmerzen, Leiden und bevorstehendem Tod verbunden sind, werden in einem gesonderten OECD-Leitfaden (8) ausführlich beschrieben. Weiteren Versuchstiergruppen können höhere oder niedrigere feste Dosen in Abhängigkeit davon verabreicht werden, ob Anzeichen von Toxizität oder Mortalität zu erkennen sind. Diese Vorgehensweise wird fortgesetzt, bis die Dosis ermittelt ist, die offensichtlich toxisch wirkt oder den Tod maximal eines Versuchstieres verursacht hat, bzw. bis bei der höchsten Dosis keine Wirkungen zu erkennen sind oder bis bereits bei der niedrigsten Dosis der Tod von Versuchstieren eintritt.
B.1.bis 1.4 Beschreibung der Prüfmethode
B.1.bis 1.4.1 Auswahl der Tierspezies
Die bevorzugte Nagetierspezies ist die Ratte, es können aber auch andere Spezies verwendet werden. In der Regel werden weibliche Tiere verwendet (7). Weibliche Tiere werden bevorzugt, da die in der Literatur zitierten Untersuchungen zu konventionellen LD50-Tests zwischen den Geschlechtern in der Regel nur geringfügige Unterschiede hinsichtlich der Empfindlichkeit belegen, in den Fällen, in denen Unterschiede beobachtet werden, jedoch zeigen, dass weibliche Tiere im Allgemeinen etwas empfindlicher sind (10). Wenn jedoch die vorhandenen Informationen über die toxikologischen oder toxikokinetischen Eigenschaften von strukturverwandten Chemikalien darauf hinweisen, dass vermutlich die männlichen Tiere empfindlicher sind, sollten männliche Tiere verwendet werden. Wird ein Versuch an männlichen Tieren durchgeführt, sollte dies hinreichend begründet werden.
Es sollen junge, gesunde, ausgewachsene Tiere aus üblicherweise eingesetzten Laborstämmen verwendet werden. Die weiblichen Tiere dürfen weder bereits geworfen haben noch momentan trächtig sein. Alle Tiere müssen zu Beginn der Verabreichung zwischen 8 und 12 Wochen alt sein; ihre Gewichtswerte müssen im Bereich von ± 20 % des mittleren Gewichts sämtlicher zuvor verwendeten Tiere liegen.
B.1.bis 1.4.2 Haltung und Fütterung
Die Temperatur des Versuchstierraums soll 22 °C (± 3 °C) betragen. Die relative Luftfeuchtigkeit soll mindestens 30 % betragen und außer während der Reinigung vorzugsweise nicht über 70 % liegen; anzustreben ist ein Wert von 50-60 %. Der Raum soll künstlich beleuchtet sein, wobei die Beleuchtung im 12-Stunden-Rhythmus ein- und ausgeschaltet werden soll. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, wobei eine unbegrenzte Trinkwasserversorgung zu gewährleisten ist. Die Tiere können in den Käfigen nach Verabreichungsdosen gruppiert werden, wobei aber die Anzahl der Tiere pro Käfig noch eine genaue Beobachtung der einzelnen Tiere ermöglichen muss.
B.1.bis 1.4.3 Vorbereitung der Versuchstiere
Die Tiere werden nach Zufallskriterien ausgewählt, zwecks Identifizierung markiert und für die Dauer von mindestens 5 Tagen vor Versuchsbeginn in ihren Käfigen gehalten, damit sie sich an die Laborbedingungen gewöhnen können.
B.1.bis 1.4.4 Vorbereitung der Dosen
Generell sollen die Testsubstanzen mit einem für den gesamten zu testenden Dosierungsbereich gleichbleibenden Volumen verabreicht werden, indem jeweils die Konzentration der Dosiszubereitung variiert wird. Wenn jedoch ein flüssiges Endprodukt oder Gemisch zu testen ist, kann die Anwendung der unverdünnten, d. h. mit gleichbleibender Konzentration verabreichten Testsubstanz für die anschließende Risikobewertung der betreffenden Substanz größere Relevanz haben und wird daher von verschiedenen Behörden vorgeschrieben. In jedem Fall darf das maximal zu verabreichende Dosisvolumen nicht überschritten werden. Welches Flüssigkeitsvolumen jeweils höchstens auf einmal verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Bei Nagetieren soll das Volumen im Normalfall 1 ml pro 100 g Körpergewicht nicht überschreiten; bei wässrigen Lösungen können aber auch 2 ml pro 100 g Körpergewicht in Betracht gezogen werden. Bezüglich der Formulierung wird, wenn dies möglich ist, eine wässrige Lösung/Suspension/Emulsion empfohlen, danach in der Reihenfolge der Präferenz eine Lösung/Suspension/Emulsion in Öl (z.B. Maisöl) sowie an dritter Stelle eventuell eine Lösung in anderen Vehikeln. Bei anderen Vehikeln als Wasser sollen die toxikologischen Eigenschaften des jeweiligen Vehikels bekannt sein. Die Dosen dürfen erst kurz vor der Verabreichung zubereitet werden, sofern die Zubereitung über den Zeitraum, in dem sie angewandt werden soll, nicht bekanntermaßen eine annehmbare Stabilität aufweist.
B.1.bis 1.5 Verfahrensweise
B.1.bis 1.5.1 Verabreichung der Dosen
Die Testsubstanz wird als Einzeldosis mit einer Sonde über einen Magensonde oder über eine geeignete Intubationskanüle verabreicht. In dem seltenen Fall, dass die Verabreichung als Einzeldosis nicht möglich ist, kann die Dosis über einen Zeitraum von maximal 24 Stunden in kleineren Teilmengen verabreicht werden.
Die Versuchstiere sollten vor der Verabreichung nüchtern sein (z.B. sollte Ratten das Futter (nicht das Wasser) über Nacht vorenthalten werden; Mäusen sollte das Futter (nicht das Wasser) 3-4 Stunden zuvor vorenthalten werden). Nach diesem Futterentzug sollen die Tiere gewogen werden und die Testsubstanz verabreicht bekommen. Nach der Verabreichung der Substanz kann das Futter bei Ratten weitere 3-4 Stunden bzw. bei Mäusen weitere 1-2 Stunden zurückgehalten werden. Wenn die Dosis über einen bestimmten Zeitraum hinweg in Teilmengen verabreicht wird, kann es je nach Länge dieses Zeitraums erforderlich sein, die Tiere mit Futter und Wasser zu versorgen.
B.1.bis 1.5.2 Vorstudie
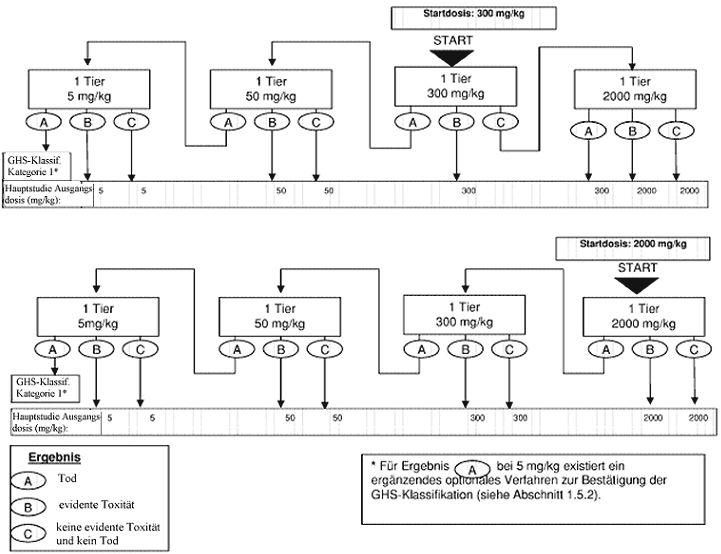
Zweck der Sichtungsstudie ist die Ermittlung der zweckmäßigen Ausgangsdosis für die Hauptuntersuchung. Die Testsubstanz wird einzelnen Tieren in einem sequentiellen Verfahren gemäß den Flussdiagrammen in Anhang 1 verabreicht. Die Sichtungsstudie ist abgeschlossen, wenn eine Entscheidung über die Ausgangsdosis für die Hauptstudie getroffen werden kann (oder wenn bereits bei der niedrigsten festen Dosis der Tod eines Versuchstiers eintritt).
Als Ausgangsdosis für die Sichtungsstudie wird von den festen Dosierungen 5, 50, 300 und 2000 mg/kg die Dosis ausgewählt, die voraussichtlich eine evidente Toxizität bewirken wird, wobei nach Möglichkeit die Erkenntnisse aus Invivo- und In-vitro-Daten für denselben chemischen Stoff sowie für strukturverwandte chemische Stoffe zugrunde liegen sollten. Wenn keine entsprechenden Informationen vorliegen, wird die Ausgangsdosis 300 mg/kg verwendet.
Zwischen den Versuchen an jedem einzelnen Tier liegt ein zeitlicher Abstand von mindestens 24 Stunden. Alle Tiere sollten mindestens 14 Tage lang beobachtet werden.
In Ausnahmefällen - und nur, wenn dies durch konkrete Vorschriften begründet ist - kann die Anwendung einer weiteren festen oberen Dosis von 5.000 mg pro kg Körpergewicht in Erwägung gezogen werden (siehe Anhang 3). Aus Tierschutzgründen wird von Tierversuchen im Bereich der GHS-Kategorie 5 (2000-5000 mg/kg) abgeraten; diese Versuche sollten nur dann in Betracht gezogen werden, wenn die Ergebnisse eines solchen Tests für den Schutz der Gesundheit von Menschen oder Tieren oder der Umwelt mit hoher Wahrscheinlichkeit direkt relevant sind.
Wenn ein Versuchstier, das mit der niedrigsten festen Dosis (5 mg/kg) getestet wird, bei der Sichtungsstudie stirbt, wird die Studie normalerweise beendet und die Substanz der GHS-Kategorie 1 zugewiesen (wie in Anhang 1 dargestellt). Ist jedoch eine weitere Bestätigung dieser Klassifizierung erforderlich, kann das folgende ergänzende Verfahren in Betracht gezogen werden: Ein zweites Versuchstier wird mit der Dosis 5 mg/kg getestet. Wenn dieses zweite Tier stirbt, wird die GHS-Kategorie 1 bestätigt und die Studie sofort beendet. Wenn das zweite Tier überlebt, wird maximal drei weiteren Tieren die Dosis 5 mg/kg verabreicht. Da ein hohes Mortalitätsrisiko besteht, sollten diese Tiere aus Tierschutzgründen nacheinander getestet werden. Der zeitliche Abstand zwischen den Versuchen an den einzelnen Tieren sollte groß genug sein, um die Überlebenswahrscheinlichkeit des vorherigen Tieres bestätigen zu können. Wenn ein zweites Tier stirbt, wird die Versuchsreihe sofort beendet, und es werden keine weiteren Tiere getestet. Da der Tod eines zweiten Tieres (unabhängig davon, wie viele Tiere bis zum Zeitpunkt der Beendigung getestet wurden) zu Ergebnis a zählt (2 oder mehr tote Tiere), wird nach der Klassifizierungsregel aus Anhang 2 für die feste Dosis 5 mg/kg vorgegangen (Kategorie 1 bei zwei oder mehr toten Versuchstieren bzw. Kategorie 2, wenn nur ein Tier stirbt). Zur Ergänzung wird in Anhang 4 die Klassifizierung im EU-System bis zur Einführung des neuen GHS erläutert.
B.1.bis 1.5.3 Hauptstudie
B.1.bis 1.5.3.1 Anzahl der Versuchstiere und Dosierung
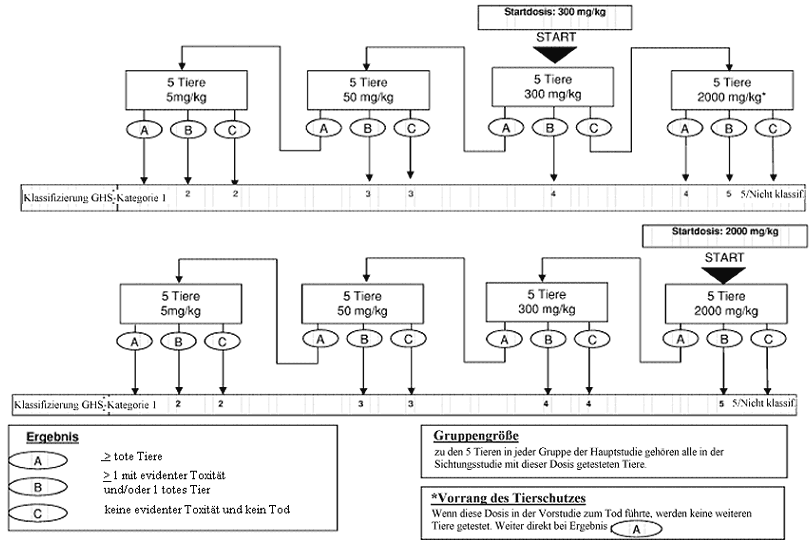
Welche Maßnahmen nach dem Versuch mit der Anfangsdosierung durchgeführt werden sollen, ist den Flussdiagrammen in Anhang 2 zu entnehmen. In Betracht kommt jeweils eine von drei Maßnahmen: Versuch beenden und die entsprechende Gefahrenklassifikationsklasse zuweisen, mit einer höheren festen Dosis testen oder mit einer niedrigen festen Dosis testen. Aus Tierschutzgründen wird jedoch eine Dosierung, die in der Sichtungsstudie zum Tod geführt hat, in der Hauptstudie nicht wiederholt (siehe Anhang 2). Die bisher gemachten Erfahrungen lassen als wahrscheinlichstes Ergebnis bei der Ausgangsdosierung vermuten, dass die Substanz ohne weitere Tests klassifiziert werden kann.
Im Normalfall werden für jede untersuchte Dosierung insgesamt fünf Tiere gleichen Geschlechts verwendet. Zu diesen fünf Tiere gehören ein Tier aus der Sichtungsstudie, dem die gewählte Dosierung verabreicht wurde, sowie vier weitere Tiere (außer wenn im Ausnahmefall eine in der Hauptstudie verwendete Dosierung in der Sichtungsstudie nicht enthalten war).
Der Zeitabstand zwischen den Versuchen mit der jeweiligen Dosierung richtet sich nach dem Einsetzen, der Dauer und dem Schweregrad der Toxizitätsanzeichen. Die Behandlung der Versuchstiere mit der nächsten Dosis sollte hinausgezögert werden, bis angenommen werden kann, dass die zuvor verwendeten Tiere überlebt haben. Bei Bedarf wird ein Abstand von drei bis vier Tagen zwischen den Versuchen mit den einzelnen Dosierungen empfohlen, um die Feststellung einer zeitverzögerten Toxizität zu ermöglichen. Dieser Zeitabstand kann nach Bedarf angepasst werden, wenn z.B. die beobachteten Reaktionen keine Schlussfolgerungen zulassen.
Wenn eine obere feste Dosis von 5.000 mg/kg in Erwägung gezogen wird, sollte verfahren werden, wie in Anhang 3 beschrieben (siehe auch Abschnitt 1.6.2).
B.1.bis 1.5.3.2 Limit-Test
Der Limit-Test wird hauptsächlich dann eingesetzt, wenn die Person, die den Test durchführt, nach ihr vorliegenden Informationen annehmen kann, dass das zu testende Material wahrscheinlich nicht toxisch ist, d.h. nur oberhalb der in Vorschriften festgelegten Grenzdosen toxisch wirkt. Informationen über die Toxizität des Testmaterials können aus Kenntnissen über ähnliche getestete Verbindungen, Gemische oder Produkte gewonnen werden, wobei Art und Prozentanteil der Komponenten zu berücksichtigen sind, deren toxikologische Relevanz bekannt ist. In den Fällen, in denen nur wenige oder keine Informationen über die Toxizität des Testmaterials vorliegen oder in denen von einer Toxizität des Testmaterials ausgegangen wird, sollte der Haupttest durchgeführt werden.
Bei der normalen Vorgehensweise wird für den Limit-Test im Rahmen dieses Leitfadens eine Ausgangsdosis von 2.000 mg/kg (bzw. im Ausnahmefall 5.000 mg/kg) für die Sichtungsstudie verwendet; anschließend werden vier weitere Tiere dieser Dosis ausgesetzt.
B.1.bis 1.6 Beobachtungen
Die Tiere werden nach der Verabreichung einzeln beobachtet: mindestens einmal während der ersten 30 Minuten, in regelmäßigen Abständen während der ersten 24 Stunden, wobei die ersten vier Stunden besonders zu beachten sind, sowie anschließend täglich während einer Gesamtdauer von 14 Tagen, sofern die Tiere nicht aus Tierschutzgründen aus dem Versuch genommen und auf humane Weise getötet werden müssen oder ihr Tod festgestellt wird. Die Beobachtungsdauer sollte nicht strikt festgelegt werden, sondern abhängig von den toxischen Reaktionen, vom Zeitpunkt des Einsetzens dieser Reaktionen und von der Länge der Erholungsdauer festgelegt werden und kann daher ausgedehnt werden, wenn dies erforderlich scheint. Der Zeitpunkt, zu dem die Anzeichen von Toxizität auftreten und wieder verschwinden, ist insbesondere dann von Bedeutung, wenn eine Tendenz zur zeitlichen Verzögerung der Toxizitätsanzeichen besteht (11). Sämtliche Beobachtungen werden für die Tiere systematisch jeweils in Einzelprotokollen dokumentiert.
Zusätzliche Beobachtungen sind erforderlich, wenn bei den Tieren weiterhin Anzeichen für Toxizität zu beobachten sind. Die Beobachtungen sollten Veränderungen an Haut und Fell, Augen und Schleimhäuten erfassen und auch die Atmung, den Kreislauf, das vegetative und zentrale Nervensystem sowie somatomotorische Aktivitäten und Verhaltensmuster berücksichtigen. Auf Symptome wie Tremores, Krämpfe, Speichelfluss, Durchfall, Lethargie, Schlaf und Koma sollte geachtet werden. Die im Humane Endpoints Guidance Document (8) zusammengefassten Prinzipien und Kriterien sind zu berücksichtigen. Tiere, bei denen ein moribunder Zustand festgestellt wird, sowie Tiere, die starke Schmerzen haben oder anhaltende Anzeichen von schwerem Leiden zeigen, sollten auf humane Weise getötet werden. Wenn Tiere aus humanen Gründen getötet werden oder ihr Tod festgestellt wird, sollte der Todeszeitpunkt so genau wie möglich registriert werden.
B.1.bis 1.6.1 Körpergewicht
Das Gewicht der einzelnen Tiere soll kurz vor der Verabreichung der Testsubstanz sowie spätestens eine Woche danach ermittelt werden. Gewichtsveränderungen sollen berechnet und registriert werden. Am Ende des Tests werden die überlebenden Tiere gewogen und anschließend auf humane Weise getötet.
B.1.bis 1.6.2 Pathologie
Alle Versuchstiere (einschließlich der Tiere, die während des Tests sterben oder aus Tierschutzgründen aus von der Studie genommen werden) sollen auf makrologische Veränderungen untersucht werden. Alle makrologischen Veränderungen sollen für jedes einzelne Tier registriert werden. Bei Tieren, die 24 oder mehr Stunden lang überlebt haben, kann auch eine mikroskopische Untersuchung von Organen mit makropathologischen Befunden in Erwägung gezogen werden, da sich hieraus eventuell nützliche Informationen gewinnen lassen.
B.1.bis 2 Daten
Es sollen Daten zu den einzelnen Tieren bereitgestellt werden. Darüber hinaus sollen sämtliche Daten in tabellarischer Form zusammengefasst werden, wobei für jede Testgruppe die folgenden Daten zu erfassen sind: Anzahl der verwendeten Tiere, Anzahl der Tiere mit Anzeichen für eine toxische Wirkung, Anzahl der Tiere, deren Tod während des Versuchs festgestellt wurde oder die aus humanen Gründen getötet wurden, Todeszeitpunkt der einzelnen Tiere, Beschreibung und Zeitverlauf der toxischen Wirkungen und ihrer Reversibilität sowie pathologische Befunde.
B.1.bis 3 Berichte
B.1.bis 3.1 Testbericht
Der Prüfbericht muss die folgenden Angaben enthalten (soweit angemessen):
Testsubstanz:
Vehikel (wenn verwendet):
Testbedingungen:
Ergebnisse:
Diskussion und Interpretation der Ergebnisse.
Schlussfolgerung.
B.1.bis 4 Literatur
(1) British Toxicology Society Working Party an Toxicity (1984). Special report a new approach to the classification of substances and preparations an the basis of their acute toxicity. Human Toxicol., 3, 85-92.
(2) Van den Heuvel, M.J., Dayan, A.D. and Shillaker, R.O. (1987). Evaluation of the BTS approach to the testing of substances and preparations for their acute toxicity. Human Toxicol., 6, 279-291.
(3) Van den Heuvel, M.J., Clark, D.G., Fielder, R.J., Koundakjian, P.P., Oliver, G.J.A., Pelling, D., Tomlinson, N.J. and Walleer, A.P. (1990). The international validation of a fixed-dose procedure as an alternative to the classical LD50 test. Fd. Chem. Toxicol. 28, 469-482.
(4) Whitehead, A. and Curnow, R.N. (1992). Statistical evaluation of the fixed-dose procedure. Fd. Chem. Toxicol., 30, 313-324.
(5) Stallard, N. and Whitehead, A. (1995). Reducing numbers in the fixed-dose procedure. Human Exptl. Toxicol. 14, 315-323. Human Exptl. Toxicol.
(6) Stallard, N., Whitehead, A. and Ridgeway, P. (2002). Statistical evaluation of the revised fixed dose procedure.-Hum. Exp. Toxicol., 21, 183 -196.
(7) OECD (2001). Guidance Document an Acute Oral Toxicity Testing. Environmental Health and Safety Monograph Series an Testing and Assessment N. 24. Paris
(8) OECD (2000). Guidance Document an the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Serien an Testing and Assesment N. 19.
(9) OECD (1998). Harmonised Integrated Hazard Classification for Human Health and Environmental Effects of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party an Chemicals in November 1998, Part 2, p.11 Ihttp://webnetl.oecd.org/oecd/pages/home/displaygeneral/0,3380,EN-documents-521-14-no-24-no-O,FF.html 1.
(10) Lipnick, R.L., Cotruvo, J.A., Hill, R.N., Bruce, R.D., Stitzel, K.A., Walker, A.P., Chu, I., Goddard, M., Segal, L., Springer, J.A. and Myers, R.C. (1995). Comparison of the Up-and-Down, Conventional LD50, and Fixed-Dose Acute Toxicity Procedures. Fd. Chem. Toxicol. 33, 223-231.
(11) Chan P.K and A.W. Hayes (1994) Chapter 16 Acute Toxicity and Eye Irritation . In: Principles and Methods of Toxicology. 3 'd Edition. A.W. Hayes, Editor. Raven Press, Ltd. New York, USA.
Anhang 1: Prüfverfahren für die Vorstudie
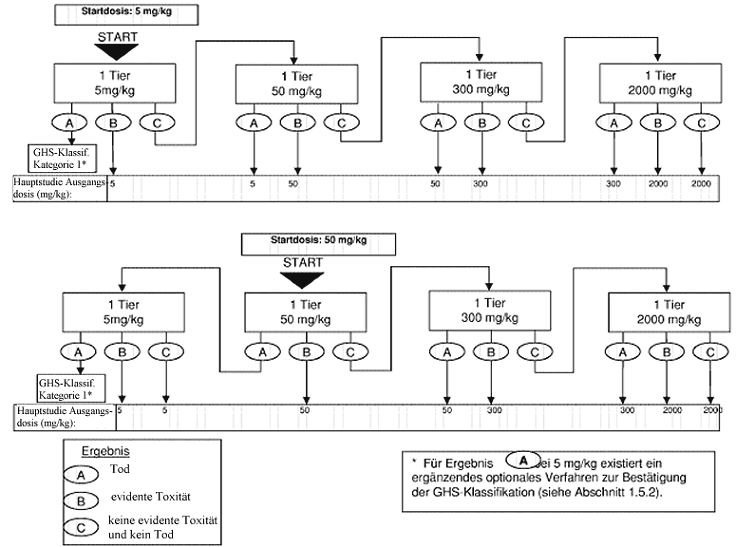
Anhang 1a: Prüfverfahren für die Vorstudie
Anhang 2: Prüfverfahren für die Hauptstudie
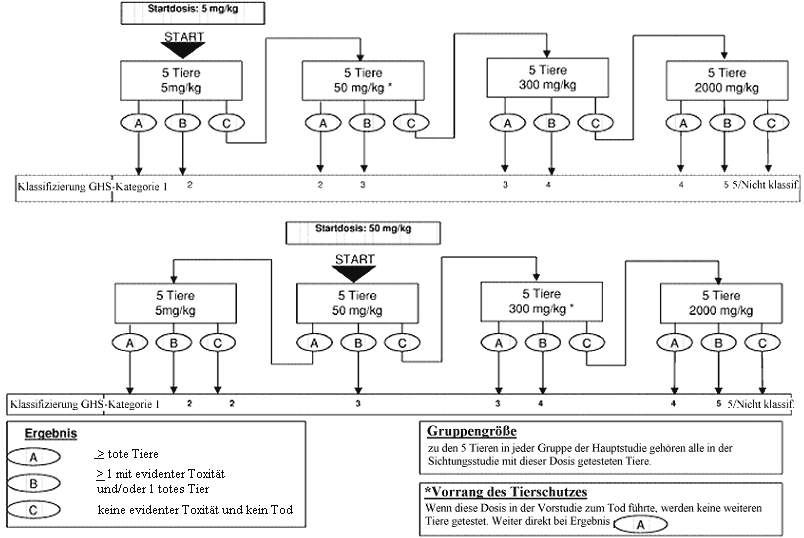
Anhang 2a: Prüfverfahren für die Hauptstudie
Anhang 3 Kriterien für die Klassifizierung von Testsubstanzen mit erwarteten LD50-Werten über 2.000 mg/kg ohne erforderliche Tests
Die Kriterien für die Gefahrenkategorie 5 sollen die Identifikation von Testsubstanzen ermöglichen, die eine vergleichsweise geringe akute Toxizitätsgefahr aufweisen, aber unter bestimmten Umständen eine Gefahr für anfällige Bevölkerungsgruppen darstellen. Bei diesen Substanzen wird von einer oralen oder dermalen LD50im Bereich 2.000-5.000 mg/kg bzw. von vergleichbaren Dosen für andere Verabreichungswege ausgegangen. Die Testsubstanzen sollte in den folgenden Fällen in die Gefahrenkategorie 2.000 mg/kg < LD50 < 5.000 mg/kg (GHS-Kategorie 5) eingestuft werden:
Versuche mit Dosen über 2.000 mg/kg
In Ausnahmefällen - und nur, wenn dies durch konkrete Vorschriften begründet ist - kann die Anwendung einer weiteren festen oberen Dosis von 5.000 mg pro kg Körpergewicht in Erwägung gezogen werden. Aus Tierschutzgründen wird von Versuchen mit 5.000 mg/kg abgeraten; diese Versuche sollen nur dann in Erwägung gezogen werden, wenn die Ergebnisse eines solchen Versuchs für den Schutz der Gesundheit von Menschen oder Tieren oder der Umwelt mit hoher Wahrscheinlichkeit direkt relevant sind (9).
Vorstudie
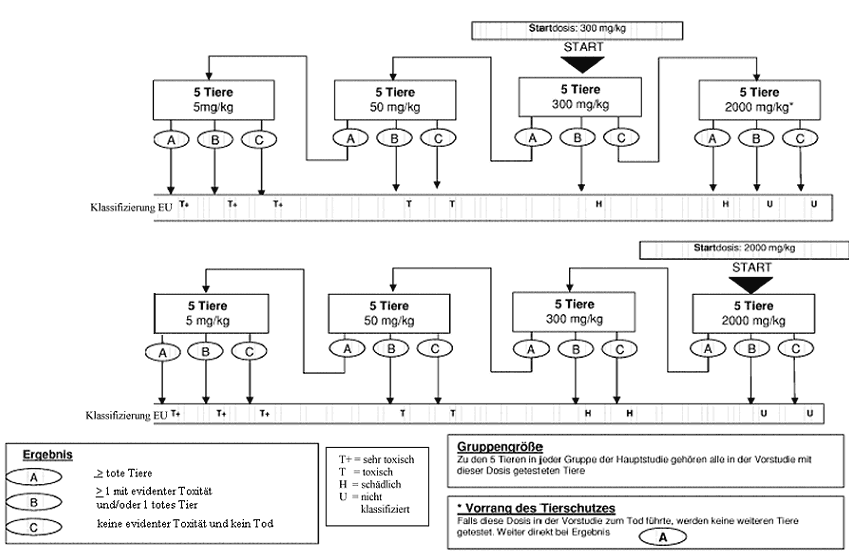
Die Entscheidungsgrundlagen für das in Anhang 1 vorgestellte sequenzielle Verfahren werden auf eine Dosis von 5.000 mg/kg erweitert: Wenn eine Startdosis von 5.000 mg/kg für die Vorstudie verwendet wird, bedingt Ergebnis a (Tod) den Test eines zweiten Tieres mit 2.000 mg/kg; die Ergebnisse B und C (evidente Toxizität oder keine Toxizität) führen zum Einsatz von 5.000 mg/kg als Startdosis für die Hauptstudie. Analog wird bei Verwendung einer anderen Startsdosis als 5.000 mg/kg der Versuch mit 5.000 mg/kg fortgesetzt, wenn bei 2.000 mg/kg Ergebnis B oder C eintritt; wenn anschließend bei 5.000 mg/kg Ergebnis a eintritt, bedingt dies eine Startdosis von 2.000 mg/kg bei der Hauptstudie, während die Ergebnisse B und C zu einer Startdosis von 5.000 mg/kg bei der Hauptstudie führen.
Hauptstudie
Die Entscheidungsgrundlagen für das in Anhang 2 vorgestellte sequenzielle Verfahren werden auf eine Dosierung von 5.000 mg/kg erweitert: Wenn in der Hauptstudie eine Startdosis von 5.000 mg/kg verwendet wird, ist bei Ergebnis a (?2 tote Versuchstiere) eine zweiten Gruppe mit 2.000 mg/kg zu testen; die Ergebnisse B (evidente Toxizität und/oder <_I totes Versuchstier) oder C (keine Toxizität) führen dazu, dass die Substanz keine Klassifizierung gemäß GHS erhält. Analog wird bei Verwendung einer anderen Startdosis als 5.000 mg/kg der Versuch mit 5.000 mg/kg fortgesetzt, wenn bei 2.000 mg/kg Ergebnis C eintritt; führt eine Gabe von 5.000 mg/kg zu Ergebnis A, wird die Substanz GHS-Kategorie 5 zugewiesen; bei den Ergebnissen B und C wird die Substanz nicht klassifiziert.
Anhang 4: Prüfmethode B.1 bis - Anleitung für die Klassifizierung gemäß dem EU-System zur Überbrückung des Übergangszeitraums bis zur vollständigen Umsetzung des "Globalen Harmonisierten Systems" (GHS) (Auszug aus Literaturhinweis (8))
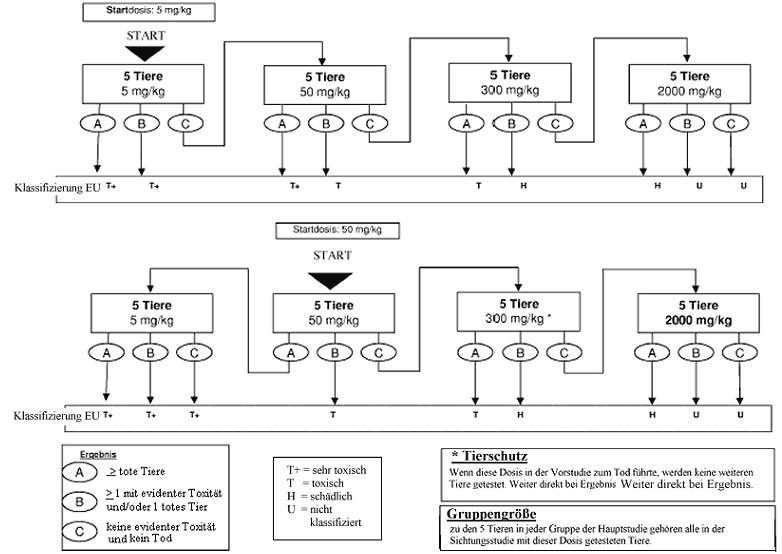
Anhang 4a: Prüfmethode B.1 bis - Anleitung für die Klassifizierung gemäß dem EU-System zur Überbrückung des Übergangszeitraums bis zur vollständigen Umsetzung des"Globalen Harmonisierten Systems" (GHS) (Auszug Literaturhinweis (8))
| |
weiter . | |
(Stand: 04.08.2022)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: ab 105.- € netto
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion