 Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk; BGI / DGUV-I

| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk; BGI / DGUV-I |
|
BGI/GUV-I 505-82 / DGUV-Information 213-582 - Verfahren zur Bestimmung von Quarz und Cristobalit
Von den Unfallversicherungsträgern anerkannte Analysenverfahren zur Feststellung der Konzentrationen krebserzeugender Arbeitsstoffe in der Luft in Arbeitsbereichen
Deutsche Gesetzliche Unfallversicherung (DGUV) Information
(Ausgabe 08/2013aufgehoben)
| Redaktioneller Hinweis: Berufsgenossenschaften sind gemäß § 210 SGB VII Behörden; ihre amtlichen Veröffentlichungen nach § 15 SGB VII unterliegen gemäß § 5 Abs. 2 UrhG keinem Urheberrechtsschutz. |
Verfahren 01
Probenahme mit Pumpe, Abscheidung der alveolengängigen Staubfraktion (A-Staub) auf Partikelfilter, Infrarotspektroskopie nach gravimetrischer A-Staubbestimmung
Quarz und Cristobalit - 01 - IR
(erstellt: 06/2012)
Verfahren 01
Probenahme mit Pumpe, Abscheidung der alveolengängigen Staubfraktion (A-Staub) auf Partikelfilter, Infrarotspektroskopie nach gravimetrischer A-Staubbestimmung
Erprobtes und von den Unfallversicherungsträgern anerkanntes Verfahren zur Bestimmung von Quarz und Cristobalit in Arbeitsbereichen.
Es sind personenbezogene und ortsfeste Probenahmen für Messungen zur Beurteilung von Arbeitsbereichen möglich.
Für die folgenden Stoffe sind Verfahren validiert.
| Name | Quarz | Cristobalit |
| CAS-Nr. | 14808-60-7 | 14464-46-1 |
| Summenformel | SiO2 | SiO2 |
| Molmasse | 60,08 g/mol | 60,08 g/mol |
Kurzfassung
Mit diesem Verfahren wird die über die Probenahmedauer gemittelte Konzentration von Quarz und Cristobalit im Arbeitsbereich personenbezogen oder ortsfest bestimmt.
| Messprinzip: | Mit Hilfe einer Pumpe wird ein definiertes Luftvolumen durch ein Partikelfilter gesaugt. Zunächst wird der A-Staub gravimetrisch und anschließend die Quarz- und Cristobalit-Konzentration infrarotspektroskopisch im A-Staub bestimmt. |
| Bestimmungsgrenze: | absolut: sie liegt für reale Staubproben erfahrungsgemäß für Quarz und Cristobalit bei rund 0,03 mg. relativ: |
| Selektivität: | Die Bestimmung ist für Quarz und Cristobalit selektiv, kann aber durch andere Staubkomponenten gestört werden. Eine eindeutige Identifizierung kann dadurch insbesondere bei niedrigen Quarz- und Cristobalit-Anteilen beeinträchtigt sein. In solchen Fällen ist gegebenenfalls auf das röntgendiffraktometrische Verfahren auszuweichen. |
| Vorteile: | Vergleichsweise einfaches Verfahren mit geringem apparativen Aufwand. Im Unterschied zur Röntgendiffraktion etwa um den Faktor 2 niedrigere Bestimmungsgrenze. |
| Nachteile: | Im Vergleich zur Röntgendiffraktion kann die Analyse stärker durch andere Staubkomponenten gestört werden. |
| Apparativer Aufwand: | Durchflussgeregelte Pumpe mit Probenahmekopf und Partikelfilter, Vakuumpumpe, Schwingmühle oder Reibschale, Glühofen, Heizplatte, Sedimentationsapparaturen bzw. fraktionierende Staubsammler (z.B. Impaktoren mit mehreren Stufen < 10 µm), Fourier-Transformations-Infrarotspektrometer (FTIR). |
1 Geräte, Materialien und Chemikalien
1.1 Geräte und Hilfsmittel für die Probenahme
1.2 Geräte für Probenvorbereitung und Bestimmung
Die in dieser Liste genannten Probengefäße werden (auch vor dem erstmaligen Gebrauch) mit destilliertem Wasser und anschließend mit Ethanol gereinigt und danach getrocknet und bei 40 - 50 °C bis zum nächsten Gebrauch gelagert.
1.3 Chemikalien
2 Probenahme
Mit Hilfe einer Pumpe wird ein definiertes Luftvolumen durch das Probenahmefilter gesaugt, das sich in dem Sammelkopf des Probenahmegerätes befindet. Mit der Probenahme wird die alveolengängige Staubfraktion (A-Staub) gemäß DIN EN 481 [ 2] erfasst. Hierfür haben sich z.B. die Probenahmesysteme PM 4F, VC 25F, FSP-10, FSP BIa und MPG II bewährt (siehe Tabelle 2 in Abschnitt 5.2). Es ist darauf zu achten, dass die Filter in den Halterungen während des Transports staubdicht verschlossen sind. Das Material der Behältnisse sollte so beschaffen sein, dass statische Aufladungen möglichst vermieden werden.
Besondere Aspekte der Probenahme von Stäuben und die Darstellung möglicher Fehlerquellen sind in [ 1] beschrieben.
3 Analytische Bestimmung
In der Regel erfolgt bei Luftproben, in denen die Konzentration silikogener Komponenten bestimmt werden soll, auch eine Bestimmung der Staubmasse zur Ermittlung der A-Staub-Konzentration. Die Kenntnis der hierdurch ermittelten Staubbelegung des Probenahmefilters ist von Bedeutung, da für die Analyse mittels Infrarotspektroskopie nur eine bestimmte Masse an Staub zur Analyse verwendet werden kann (bis zu 2 mg).
3.1 Probenaufbereitung
Im Folgenden werden zwei Varianten der Aufbereitung der Proben beschrieben. Durch Vorversuche ist zu ermitteln, welches Verfahren für die Anwendung geeignet ist.
Bei Anwendung der Variante 1 kann es z.B. bei zu schneller Aufheizrate bei der Vorbehandlung auf der Heizplatte zu nicht reproduzierbaren Minderbefunden kommen. Als Grund hierfür wird eine Reaktion zwischen Filtermaterial, Kaliumbromid (KBr) und Staub bei der Aufbereitung vermutet: Bei zu schnellem Aufheizen des beaufschlagten Nitrocellulosefilters in Anwesenheit von KBr kann Kaliumnitrat bzw. -nitrit und Kaliumoxid entstehen, das durch nachfolgende Reaktion mit dem Quarz der Probe Silikate bilden kann (Bildung von KNO2 aus KNO3 ab 330 °C, daraus K2O und nitrose Gase ab 441 °C). Hierdurch wird der zu bestimmende Quarz der Probe selektiv umgewandelt. Sollte bei eigenen Versuchen ein Minderbefund an Quarz festzustellen sein, ist eine langsamere Aufheizrate zu wählen oder gemäß Variante 2 vorzugehen. Vergleichbare Effekte sind auch für die Aufbereitung von Cristobalit- haltigem Staub zu überprüfen.
Für die im Folgenden beschriebenen Aufbereitungsverfahren gilt allgemein, dass eine Presstablette nicht mehr als 2 mg Staub und nicht mehr als 1 mg Quarz enthalten soll. Bei Verwendung von bis zu 2 mg Staub ist zumeist ein Quarzgehalt von deutlich unter 1 mg im Staub zu erwarten, da in der überwiegenden Zahl von Arbeitsbereichen Quarzgehalte von unter 20 % Quarz auftreten. In bestimmten Bereichen (z.B. Aufbereitung von Quarzsand oder -mehl) können aber auch Quarzgehalte im Staub von mehr als 50 % angetroffen werden. Die für die Herstellung einer Presstablette verwendete KBr-Menge variiert je nach Gerätekonfiguration zwischen etwa 250 und 450 mg. Die Massen der Presstabletten der Kalibrierung als auch der Proben sollen nicht mehr als 10 % voneinander abweichen.
Es empfiehlt sich, durch Vorversuche die maximal zu verwendende Staubmasse in einer Presstablette durch Versuche zu ermitteln. Eine zu große Staubmasse in einer Presstablette führt zu einer Reduzierung der Transmission, welche wiederum zu einer schlechteren Bestimmungsgrenze führt. In Einzelfällen führen auch bestimmte Begleitkomponenten einiger Arbeitsplatzstäube schon bei einer Masse von weniger als 1 mg zu Störungen der Transmission.
Variante 1
Filterveraschung bei Staubmassen unter 5 mg
Bei Staubmassen unter 5 mg werden die Filter unter Zusatz von 250 - 450 mg KBr verascht. In folgenden Fällen kann hiervon abgewichen werden:
Das Filter wird mit einem Skalpell oder einer Schere halbiert und mit den staubbeaufschlagten Seiten aufeinander gelegt. In einem flachen konditionierten Porzellantiegel (zuvor geglüht bei 550 °C, im Exsikkator abgekühlt, gewogen) wird eine Spatelspitze KBr verteilt, die Filterhälften auf das KBr-Bett gelegt und die Restmenge des KBr (Gesamtmasse 250 - 450 mg) auf der Oberseite der Filterhälften verteilt.
Der Tiegel wird im Abzug auf eine kalte Heizplatte gestellt und die Heizstufe so eingestellt, dass die Heizplattentemperatur bei dieser Einstellung bis auf ca. 220 °C steigt. Es wird so lange erhitzt, bis das Filter vollständig schwarzbraun erscheint (ca. 2 h, längeres Erhitzen ist unschädlich).
Danach wird der Tiegel im Glühofen aufgeheizt und bei 550 °C geglüht (1 - 1,5 h Aufheizung, 2,5 - 3 h Haltezeit). Nach Öffnen des Ofens wird der abgekühlte, aber noch leicht warme Tiegel in einen Exsikkator mit Silicagel als Trockenmittel überführt, ohne Vakuum anzulegen. Nach Erkalten des Tiegels wird der Inhalt mit Hilfe eines Spatels und eines Pinsels in eine Reibschale überführt, mit dem Pistill unter leichtem Druck homogen verrieben und mit Hilfe von Spatel und Pinsel in das Presswerkzeug zur Herstellung der Presstablette überführt.
Filterveraschung bei Staubmassen über 5 mg
Das Filter wird in der Mitte so gefaltet, dass die staubbeaufschlagten Hälften aufeinander liegen, in einen zuvor konditionierten und gewogenen Porzellantiegel gelegt und mit 1,3-Butandiol satt getränkt. Die Probe im Tiegel wird wie oben beschrieben auf der Heizplatte vorverascht und dann im Glühofen bei 550 °C geglüht (1 - 1,5 h Aufheizung, 2,5 - 3 h Haltezeit). Die Tiegel werden im Exsikkator erkalten gelassen und der Glührückstand bestimmt. Der Glührückstand wird mit Hilfe eines Spatels und des Pinsels im Tiegel zusammengeschoben und ein exakt eingewogenes Aliquot im Bereich von 0,5 - 2,5 mg in die Reibschale überführt (wenn Informationen über zu erwartende hohe Quarz- bzw. Cristobalit-Gehalte vorliegen, sind geringere Einwaagen sinnvoll). Der Glührückstand wird mit ca. 250 - 450 mg KBr versetzt, mit dem Pistill unter leichtem Druck homogen verrieben und mit Hilfe von Spatel und Pinsel in das Presswerkzeug überführt.
Variante 2
Das mit der A-Staub-Fraktion beaufschlagte Membranfilter wird in einem zuvor konditionierten und gewogenen Porzellantiegel mit 1,3-Butandiol getränkt (bis zu 3 ml) und zur Entfernung des Filtermaterials auf der Heizplatte stufenweise erhitzt:
Es verbleibt ein schwarzer Rückstand. Anschließend wird die verbleibende Probe im Glühofen langsam bis auf 600 °C erhitzt (ca. 1 - 2 Stunden Aufheizzeit). Die Veraschung dauert insgesamt ca. 6 Stunden und soll eine Störung der IR-Bestimmung verhindern. Nach dem Abkühlen wird zum Glührückstand (= quarzhaltiger A-Staub) eine definierte Menge an KBr gegeben. Auf je maximal 2 mg Staub kommen 250 - 450 mg KBr. Zum restlosen Ablösen der Substanz von der Tiegelwandung wird der Tiegel mit Staub und KBr 15 Minuten im Ultraschall behandelt. Nach der Überführung in einen Metallbecher der Mühle wird die Probenmischung 5 Minuten lang durch Mahlen homogenisiert. Längere Mahldauern bringen keine Verbesserung.
Herstellen der Presstabletten
Von der Mischung aus Probensubstanz und KBr (Probenmischung) wird die für die Herstellung des Presslings benötigte KBr-Menge eingewogen (genaue Bestimmung durch Wägung, typischer Presslingsdurchmesser: 13 mm). Beim Einfüllen der Mischung in das Presswerkzeug ist auf eine homogene Verteilung zu achten, um Massenanhäufungen beim Pressen zu vermeiden. Hierdurch soll eine möglichst gleichmäßige Dicke des Presslings erreicht werden. Nach Einbringen des Presswerkzeuges in die hydraulische Presse wird das Presswerkzeug ca. 1 Minute mit der Vakuumpumpe entgast und die KBr-tablette mit einem Druck von 80 kN/cm2 ca. 1 Minute gepresst.
Sichtbare Eintrübungen am Rand des fertigen Presslings sind ein Hinweis auf einen nicht optimalen Pressvorgang mit ungleich verteilter Mischung. Eine Überprüfung des Pressvorgangs und der Presseinrichtung kann erfolgen, indem Presslinge jeweils viermal gemessen werden, nachdem sie in der Halterung jeweils um 90 Grad in dieselbe Richtung rotiert wurden. Weichen die Ergebnisse signifikant voneinander ab, muss der Pressvorgang optimiert werden.
Um generell eine Trübung der Presstabletten zu vermeiden, sind die Probenmischung, das KBr, die Mahlgefäße und Presswerkzeuge bei 40 °C im Trockenschrank zu trocknen und zwischen den Pressprozessen aufzubewahren. Werden Presstabletten nicht unmittelbar analysiert, können diese kurzfristig im Trockenschrank gelagert werden. Nach längerer Aufbewahrung müssen die Presstabletten neu gemahlen und gepresst werden (Masseverluste berücksichtigen, tablette vor und nach dem Neupressen wiegen!).
Herstellen von Kalibrierstandards
Zur Kalibrierung ist die Verwendung zertifizierter Referenzmaterialien (auch diese enthalten in der A-Staub-Fraktion amorphes SiO2) nicht notwendig. Als Referenzmaterial sollten Quarzstäube verwendet werden, die den im Arbeitsbereich eingesetzten oder auftretenden Quarzstäuben vergleichbar sind. Eine Reinheit von z.B. 97 % Quarz, also kristallinem Anteil, ist durchaus ausreichend. Der Anteil anderer Phasen im gewählten Standard sollte unter 5 % liegen und kann phasenkontrastmikroskopisch abgeschätzt werden (z.B. Anwendung des Analysenverfahrens nach BGI 505-30 [ 9]; Eugenol als Einbettungsflüssigkeit zur Unterscheidung von Quarz und anderen Mineralphasen). Technisch verwendete Cristobalit-Stäube erreichen nicht die genannte Reinheit, typische Cristobalit-Anteile liegen bei ca. 70 % (häufig neben Quarz und amorpher Kieselsäure). Der mikroskopisch abgeschätzte Cristobalit-Anteil wird bei der Kalibrierung verwendet; alternativ können NaOH-behandelte Cristobalit-A-Stäube eingesetzt werden, deren Quarzgehalt röntgendiffraktometrisch bestimmt wurde [ 7].
Presslinge für Kalibrierreihen sind den gleichen Aufbereitungsschritten zu unterziehen, denen auch die Proben unterliegen, um anhand dieser Kalibrierstandards die Bestimmungsgrenze des Verfahrens realistisch abschätzen zu können.
Für die Zugabe der definierten Masse des Quarz- oder Cristobalit-Standards einer Kalibrierprobe haben sich zwei Methoden bewährt:
Es empfiehlt sich, zwei Kalibrierreihen zu erstellen. Eine Kalibrierung sollte dabei den gesamten Anwendungsbereich des Verfahrens abdecken (maximal 1 mg) und aus mindestens 6, besser aber 10 äquidistanten Konzentrationsstufen zwischen 0,1 und 1 mg bestehen (jeweils Doppelbestimmung). Hierdurch kann die Linearität der Kalibrierung im gesamten Auswertebereich kontrolliert und belegt werden. Um die Nachweisgrenze des Verfahrens abschätzen zu können und die Vorgehensweise bei der Auswertung zu optimieren (zu Auswerteproblemen siehe Abschnitt 4), sollte eine zweite Kalibrierung im Bereich zwischen 0,01 und 0,1 mg Quarz bzw. Cristobalit erstellt werden, ebenfalls aus mindestens 6 äquidistanten Konzentrationsstufen.
3.2 Analyse
Am Standort des FTIR-Gerätes sollte während der Messung die Raumtemperatur stabil bleiben und Zugluft sowie Sonneneinstrahlung vermieden werden. Auch eine Einwirkung durch fremde Vibrationen (z.B. Geräte in der Nachbarschaft) auf das Instrumentarium des Strahlengangs stört die Aufnahme eines Spektrums und erhöht unkontrolliert den Untergrund (Rauschen der Basislinie).
Den Gerätebeschreibungen des Herstellers ist zu entnehmen, unter welchen Bedingungen der Betrieb zu erfolgen hat; z.B. kann es notwendig sein, die Messkammer mit getrockneter Luft zu spülen oder Trockenpatronen zu verwenden, um eine Eintrübung bestimmter Elemente des optischen Systems zu vermeiden.
Die optimale Zahl der Scans, die bei der Analyse einer Probe durchgeführt werden, ist durch Vorversuche zu ermitteln. Zur Auswahl der Apodisation kann auf Empfehlungen des Geräteherstellers verwiesen bzw. durch Vorversuche ein für die Anwendung optimales Verfahren gewählt werden. Für die Analysen gemäß dieser Methode ist eine spektrale Auflösung von 4 cm-1 ausreichend (Parameterbeispiel für Bruker Vector 22: spektrale Auflösung 4 cm-1, Bereich 4000 cm-1 bis 350 cm-1, Apodisation: Blackman-Harris-3-Term, Zero-Filling Faktor 4, 120 Scans, Forward-Backward-Betrieb, Phasenkorrektur: Mertz-Methode, Phasenauflösung: 128 Punkte).
4 Auswertung
Die Auswertung des FTIR-Spektrums zur quantitativen Bestimmung der Quarzmasse erfolgt im Bereich der Doppelbande bei den Wellenzahlen 798 cm-1 und 779 cm-1, die sich über den Bereich von 850 bis 720 cm-1 erstreckt (siehe Abbildung 1). Die Bestimmung der Cristobalit-Masse erfolgt anhand der Bande bei der Wellenzahl 621,5 cm-1 (siehe Abbildung 7).
Bei der Auswertung sind zwei wesentliche Aspekte zu bedenken:
Zur Auswertung stehen die drei folgenden Verfahren zur Verfügung:
Die Auswertung muss mindestens aus einer kombinierten Anwendung der Auswerteverfahren mittels integraler Absorption (Abschnitt 4.1) und Subtraktion von Referenzspektren (Abschnitt 4.2) bestehen. Die Anwendung des Verfahrens zur Berücksichtigung des Korngrößeneinflusses (Abschnitt 4.3) ist optional und wird vor allem dann empfohlen, wenn eine vom eingesetzten Standard deutlich abweichende Korngrößenverteilung der eingesetzten Materialien oder auftretenden Stäube im Bereich der alveolengängigen Fraktion zu vermuten ist (z.B. Erzbergbau, Kohlebergbau, Einsatz von Ultrafeinstmehlen mit medianem Korndurchmesser von 1 µm). Die Korngrößenverteilung ist im FTIR-Spektrum nicht zu erkennen, daher sollten Informationen, z.B. aus Sicherheitsdatenblättern oder technischen Informationen, herangezogen werden.
Im Folgenden werden die drei Verfahren im Detail beschrieben, wobei die Vorzüge und Grenzen sowie zu erwartende Fehlergrößen bei der Anwendung erläutert werden.
Um die drei Auswerteverfahren und deren Fehler besser einschätzen zu können, empfiehlt es sich, Mischproben herzustellen, die neben einem Standard-Quarz/ -Cristobalit unterschiedliche Anteile von Störkomponenten enthalten (z.B. TiO2, CaCO3, Korund, Feldspäte, Sillimanit). Die Mischproben eignen sich für Übungszwecke und schulen im Umgang mit störenden Einflüssen bei der Quantifizierung. Außerdem können somit zu erwartende Störungen bei bekannter Staubzusammensetzung besser erkannt und die Auswertestrategie danach optimiert werden. Für die Optimierung der Auswertung von Proben mit nur geringen Anteilen an Quarz bzw. Cristobalit ist eine Kalibrierreihe im unteren Anwendungsbereich geeignet (siehe Abschnitt 3.1).
Die Auswahl eines geeigneten Standards für die Kalibrierung ist nicht unproblematisch. Grundsätzlich muss der verwendete Standard auf Grund des starken Korngrößeneinflusses eine Korngrößenverteilung der alveolengängigen Staubfraktion gemäß DIN EN 481 [ 2] aufweisen. Es sollte sich bei dem Standard jedoch nicht um einen idealen Quarz im Sinne eines gemahlenen Einkristalls handeln. Es ist bekannt, dass Proben von gezüchteten, kristallographisch nahezu störungsfreien Einkristallen bei FTIR-Analysen höhere integrale Intensität aufweisen als in Arbeitsbereichen auftretender Quarz. Eine Verwendung solcher idealer Quarze hätte zur Folge, dass die Quarzkonzentrationen an Arbeitsplätzen unterbewertet würden. Es wird deshalb empfohlen, als Standard zur Kalibrierung solche Quarzstäube zu verwenden, die auch industriell eingesetzt werden. Dies sind z.B. Sikron SF 600 der Fa. Quarzwerke, Frechen, oder Minusil 5 der Fa. U.S. Silica (USA). Es sollte auf jeden Fall eine mikroskopische oder laserbeugungsspektroskopische Kontrolle der Korngrößenverteilung erfolgen. Eventuelle Anteile gröberer Partikel sind durch Sichtung oder Sedimentation zu entfernen. Erläuterungen zur Auswahl und Aufbereitung von Quarz- und Cristobalit-Proben für die Verwendung als Standard finden sich auch in Abschnitt 4.3.
4.1 Auswertung durch Bestimmung der integralen Absorption
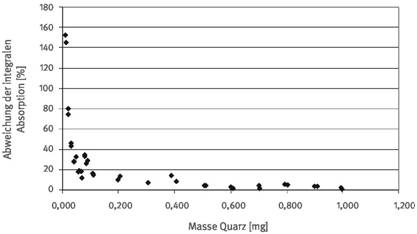
Die Bestimmung der Quarz- bzw. Cristobalit-Masse einer Staubprobe mittels integraler Absorption im Bereich der auszuwertenden Doppelbande bzw. Bande ist die einfachste Auswertemethode. Abbildung 1 zeigt diese beispielhaft für Quarz (Absorptionsspektrum eines reinen Quarzes, nicht durch andere Staubkomponenten beeinträchtigt). Allerdings weist dieses Verfahren bei Störungen durch andere Begleitkomponenten im zu analysierenden Staub auch die größten Fehler auf. Die wesentlichen Probleme bei der Ermittlung der integralen Absorption sind Störungen in den Flanken durch Absorptionsbanden anderer Phasen und eine nicht ebene Basislinie im Bereich der Integration durch mulden- bzw. buckelförmige Störungen. Störungen in den Flanken führen dazu, dass die Integrationsgrenzen nicht eindeutig festgelegt werden können und die zu integrierende Fläche durch Überlagerungen nicht systematisch falsch positiv oder negativ ermittelt werden kann. Ebenso problematisch ist die Bestimmung der integralen Absorption bei buckelförmigen (falsch positiver Befund) oder muldenförmigen Störungen der Basislinie (falsch negativer Befund; siehe Abbildung 2). Der Einfluss des Basislinienverlaufs macht sich vor allem bei niedrigen Quarz- bzw. Cristobalit-Konzentrationen bemerkbar (siehe Abbildung 3). Bei Quarzmassen unter ca. 100 µg sind Fehler von mehr als 50 % relativ zu erwarten.
Abb. 1 Absorptionsspektrum von Quarz im Bereich der Doppelbande bei den Wellenzahlen 798 cm-1 und 779 cm-1. Die Markierungen bei Wellenzahl 850 cm-1 und 720 cm-1 zeigen die Integrationsgrenzen.

Abb. 2 Absorptionsspektrum von Quarz (80 µg) im Bereich der Doppelbande mit ebener Basislinie (schwarz) und muldenförmiger Basislinie (grau). Die rechte Begrenzung der zu integrierenden Fläche der Doppelbande ist von 720 cm-1 scheinbar auf etwa 765 cm-1 bei der Muldenlage verschoben.
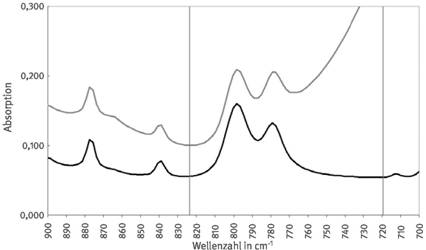
Abb. 3a R elativer Fehler bei der Bestimmung der Quarzmasse mittels Bestimmung der integralen Absorption bei Vorliegen buckelförmiger Störungen der Basislinie
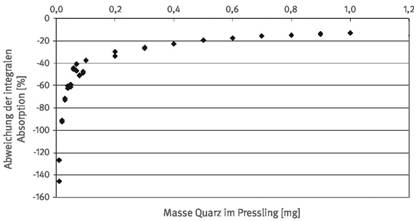
Abb. 3b Relativer Fehler bei der Bestimmung der Quarzmasse mittels Bestimmung der integralen Absorption bei Vorliegen muldenförmiger Störungen der Basislinie
Eine Auswertung allein anhand der Bestimmung der integralen Absorption sollte nicht erfolgen. Es wird empfohlen, dieses Verfahren nur in Kombination mit den Verfahren nach Abschnitt 4.2 und/oder Abschnitt 4.3 anzuwenden, um mögliche Störungen identifizieren zu können. Aufschluss über den tatsächlichen Verlauf der Basislinie kann die Spektrensubtraktion nach Abschnitt 4.2 liefern.
Generell bleibt noch festzustellen, dass bei Ermittlung der Quarz- oder Cristobalit-Masse mittels integraler Absorption der Korngrößeneinfluss (siehe Abschnitt 4.3) nicht berücksichtigt wird. Im Extremfall (Staubfraktionen mit medianem Durchmesser < 0,8 µm oder > 4 µm) können hierdurch relative Fehler von bis zu ± 20 % auftreten. Bei den häufig auftretenden Kornverteilungen Quarz-/Cristobalithaltiger Stäube an Arbeitsplätzen ist von Fehlern von bis zu ± 10 % auszugehen.
4.2 Auswertung durch Subtraktion eines Referenzspektrums
Die Bestimmung der Quarz- oder Cristobalit-Masse kann auch durch Subtraktion des Spektrums eines reinen Standards mit definierter Einwaage vom Absorptionsspektrum der zu analysierenden Probe erfolgen. Die Subtraktion wird für den Auswertebereich der Doppelbande bzw. Bande durchgeführt, der auch zur Bestimmung der integralen Intensität verwendet wird (siehe Abschnitt 4.1). Der Faktor der Subtraktion wird so gewählt, dass das verbleibende Subtraktionsergebnis einen möglichst plausiblen Basislinienverlauf annimmt. Hierbei empfiehlt es sich, den Subtraktionsfaktor um den gewählten Faktor zu variieren, um erkennen zu können, wann die Subtraktion noch nicht ausreichend oder schon zu groß ist. Da unterschiedliche Quarze oder Cristobalite nicht immer eine völlig identische Morphologie der Absorptionsbanden aufweisen, sollten nach Möglichkeit Standardspektren von verschiedenen verfügbaren Quarz- oder Cristobalit-A-Stäuben mit bekannter Reinheit aufgenommen und zur Spektrensubtraktion verwendet werden. In Abbildung 4 ist das Ergebnis einer Spektrensubstitution für eine quarzhaltige Probe beispielhaft dargestellt. Da die Lage der beiden Absorptionsmaxima der Doppelbande bei der Probe und dem Standard nicht exakt übereinstimmen, ergibt sich als verbleibende Basislinie (in Abbildung 4 überhöht dargestellt) eine doppelt geschwungene Wellenlinie. In diesem Fall sollte ein anderer Quarzstandard verwendet werden, um den Faktor der Subtraktion optimal ermitteln zu können.
Abb. 4 Bestimmung der Quarzmasse einer Staubprobe durch Subtraktion des Spektrums eines Quarzstandards vom Absorptionsspektrum der Probe im Bereich der Doppelbande (Erläuterungen siehe Text).
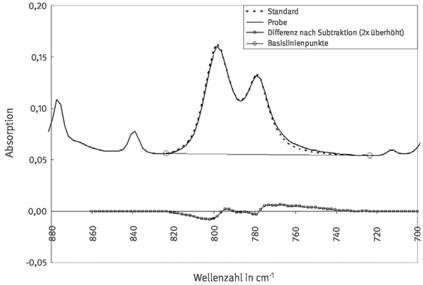
Der Vorteil bei der Anwendung dieses Auswerteverfahrens im Vergleich zur Bestimmung der integralen Absorption gemäß Abschnitt 4.1 liegt darin, dass auch bei stark gestörten Basislinienverläufen, einschließlich buckel- und muldenförmiger Basislinien und Flankenlagen der Absorptionsbanden, zumeist befriedigende Quantifizierungen durchgeführt werden können. Ergibt die Anwendung dieses Verfahrens keinen zufriedenstellenden (plausiblen) Basislinienverlauf des Subtraktionsergebnisses, ist auf das röntgendiffraktometrische Analysenverfahren auszuweichen. Eine verlässliche Quantifizierung der Quarz- oder Cristobalit-Masse ist dann mittels Infrarotspektroskopie nicht möglich.
Wie im Verfahren nach Abschnitt 4.1 wird auch hier nicht der Korngrößeneinfluss der zu analysierenden Proben berücksichtigt.
4.3 Auswertung unter Berücksichtigung des Korngrößeneinflusses
Quarz
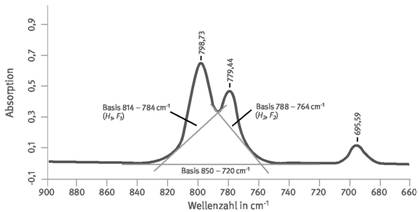
Die analytischen Absorptionsbanden 798 und 779 cm-1 stehen unter einem störenden Einfluss von vielen Mineralen, wenn zur quantitativen Bestimmung der Peakflächen (F) die Basislinie 850 - 720 cm-1benutzt wird, welche sich über einen weiten Bereich ausdehnt und eventuell in die Anstiegsbereiche anderer Mineralbanden hineinreicht (siehe Abschnitt 4.1). Deshalb wird bei dem im Folgenden beschriebenen Auswerteverfahren mit engeren Basislinienbereichen gearbeitet und die Doppelbande mit den Basislinien 814 - 784 und 788 - 764 cm-1, wie in Abbildung 5 dar gestellt, benutzt [ 3, 4].
Abb. 5 IR-Spektrum von Quarz mit Darstellung der Basislinien zur Integration der Quarzdoppelbande; H: Lot Peakspitze-Basislinie, F: Integral (Fläche) über Basislinie in Extinktion (Absorption).
In die Untersuchungen wurden industriell hergestellte Quarzmehle aus Sanden und gemahlene Bergkristalle verschiedener Herkunft einbezogen.
Die Quarzbanden sind auch im Partikelgrößenbereich von A-Staub sehr partikelgrößenabhängig; so kann die partikelgrößenbedingte Extinktionsabweichung bei Wahl eines nicht der Partikelgröße in der Probe angepassten Quarzstandards im Extremfall zu relativen Fehlern der Quarzbestimmung von bis zu ± 20 % führen (bezogen auf die Bestimmung der integralen Absorption). Daher wurde eine Funktion ermittelt, die bei Auftragen der auf die Quarzmasse m = 1 mg normierten Extinktionen H (Peakhöhe über Basislinie) und F (Peakfläche) mit (H1 * H2)1/2/mreal gegen ((H1 * H2)/ (F1 * F2))1/2 von Quarzen verschiedener Herkunft und Partikelgrößenverteilung (innerhalb der A-Staubfraktion) einen linearen Zusammenhang ergibt (siehe Formel (1)). Hiermit können die Quarzmassen m partikelgrößenunabhängig bestimmt werden (siehe Abbildung 6).
| (1) | 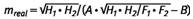 |
| mreal = | analysierte Masse von Quarz in der Probe [mg] |
| H1, H2 = | Bandenhöhe Peak 1 und 2 [Absorption des Peakmaximums] |
| F1, F2 = | Flächenwert Peak 1 und 2 [integrale Absorption der Peakfläche] |
| A = | Steigung der Regressionsgerade (siehe unten) |
| B = | Achsenabschnitt der Regressionsgerade (siehe unten) |
Die Quarzmasse m sollte 1 mg nicht überschreiten.
Die Koeffizienten a (24,218) und B (1,585) berechnen sich aus der Regressionsgeraden der Kalibrierung (Abbildung 6). Sie sind geräte- und softwarespezifisch (Integrationsparameter) und können nicht auf andere IR-Geräte übertragen werden; verwendet wurde hier das Bruker Vector 22 mit der Software OPUS 6.
Abb. 6 Zusammenhänge zwischen Höhen- (H) und Flächenwerten (F) verschiedener Quarze unterschiedlicher Korngrößenverteilungen.

Voraussetzung für die Ermittlung der notwendigen Parameter zur Anwendung dieses Auswerteverfahrens ist die Analyse unterschiedlicher Quarzfraktionen mit verschiedenem Median der Kornverteilung zwischen etwa 1,5 und 4 µm. Um möglichst enge Kornverteilungen zu erreichen, können verschiedene Kornfraktionen eines Materials z.B. mittels Kaskadenimpaktor oder Sedimentationsverfahren getrennt werden.
Neben der Herstellung eigener Quarzstaub-Fraktionen aus industriell eingesetzten Quarzen kann Quarz unterschiedlicher Herkunft, z.B. gemahlener Bergkristall, verwendet werden.
Die Gewinnung eines Quarz-A-Staubstandards aus einem Quarzmehl, z.B. Dörentruper Quarz Nr. 12 (DQ12) oder Sikron SF 600 der Fa. Quarzwerke, Frechen, kann auch durch Aufwirbeln des Quarzmehls mittels eines leichten Luftstromes in einem Gefäß, an das ein Feinstaub-Probenahmegerät MPG II angeschlossen ist, erfolgen (siehe z.B. [ 10]).
In Partikelgrößenfraktionen unterhalb ca. 1,5 µm enthält der Quarz, bedingt durch Aufmahlprozesse, deutliche Gehalte an amorphem SiO2. Für Kalibrierzwecke müssen sie daher durch eine Natronlaugebehandlung nach Baumann [ 5, 6] gereinigt werden (Auflösen des amorphen SiO2). Für die Ableitung der Koeffizienten a und B ist eine Quarzstaub-Fraktion mit mittlerer Korngröße von unter 1,5 µm aber nicht zwingend notwendig.
Eine Kalibrierfunktion ohne Korrektur des Partikelgrößeneinflusses ergibt sich durch Auftragung unterschiedlicher Massen eines Standards definierter Korngrößenverteilung gegen die Extinktionen, so z.B. gegen die geometrischen Mittelwerte vonH1 undH2:
| (2) | 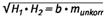 |
b = Steigung der Kalibrierfunktion
Hier wird ein Quarz-A-Staubstandard (z.B. DQ12 A-Staub) mit einem Mediandurchmesser von ca. 2,2 µm verwendet, der sich im mittleren Bereich der Funktion in Abbildung 6 befindet. Der Faktor F wird definiert alsmunkorr/mreal. Er gibt die Abweichung der Masse einer Probe bei einer Auswertung mittels einer Kalibrierung ohne Berücksichtigung der Partikelgrößenkorrektur wieder. Der Faktor F ergibt sich zu
| (3) | 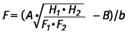 |
Der FaktorF ist 1, wenn die Extinktionen des verwendeten Quarz-A-Staubstandards eingesetzt werden. Die Faktoren F werden für die Endpunkte der Funktion in Abbildung 6 berechnet (gröbste und feinste Partikelfraktion); damit ergeben sich für den Gültigkeitsbereich der Berechnungsformel Werte zwischen F = 0,6 für die gröbsten Partikel (Median der Kornverteilung 4 µm) und F = 1,4 für die feinsten Partikel (Median der Kornverteilung 0,8 µm). Außerhalb dieser Grenzen ist diese Auswertung nicht anwendbar. Der Faktor F muss somit bei jeder Analyse mit bestimmt werden.
Es ist notwendig, für das jeweils verwendete FTIR-Analysensystem die Berechnungsformel mit Hilfe von partikelgrößenklassierten Quarzstaub-Fraktionen individuell zu erstellen.
Hinweis: Die Lagerung dieser Presslinge kann zu Eintrübungen der KBr-tabletten führen, die abweichende Messergebnisse zur Folge haben. Die KBr-tabletten müssen in diesem Fall vor der Messung gemahlen und neu gepresst werden.
Bei der Quarzanalyse muss jedes Probenspektrum daraufhin überprüft werden, ob die Basislinien Über- bzw. Unterschneidungen in den Flanken der Banden aufweisen. Zur Festlegung der Basislinienpunkte siehe Abbildung 5 und den dazu erläuternden Text. Ist dies der Fall, sollte versucht werden, ob eine Basislinienkorrektur unter Verwendung des gesamten Spektrums mit möglichst vielen Stützstellen Abhilfe schafft. Hierfür bietet die mit den FTIR-Geräten gelieferte Auswertesoftware in der Regel geeignete Möglichkeiten. Die Durchführung einer Basislinienkorrektur erfordert Erfahrung. Sie muss vorab an Beispielspektren mit bekannten Störkomponenten gewonnen werden.
Cristobalit
Ohne Korrektur des Partikelgrößeneffekts sind Fehlbestimmungen der Cristobalit-Masse anhand der Bande bei 621,5 cm-1 von maximal ± 15 % (integrale Extinktion F) möglich. Zur Bestimmung der Cristobalit-Konzentration in A-Stäuben wurde analog zum Quarz eine Formel abgeleitet, welche die Korrektur des Christiansen-Effektes beinhaltet [ 7]. Sie stellt die Abhängigkeit der Extinktion (Höhen- und Flächenwerte) unterschiedlicher Cristobalit-Proben von ihren mittleren Partikelgrößen und gleichzeitig von der Konzentration dar. Für Cristobalit vereinfacht sich die Formel (1) zur folgenden Formel (4), da nur jeweils eine Absorptionsbande ausgewertet wird:
| (4) |  |
| mreal | = analysierte Masse von Cristobalit in der Probe [mg] |
| H | = Bandenhöhe [Absorption des Peakmaximums] |
| F | = Flächenwert [integrale Absorption der Peakfläche] |
| A | = Steigung der Regressionsgerade |
| B | = Achsenabschnitt der Regressionsgerade |
Die Untersuchungen an fraktioniertem Cristobalit SF 6000, SF 3000 bzw. Sepasil (Fa. Quarzwerke, Frechen) haben gezeigt, dass diese Formel für den Bereich der mittleren Partikeldurchmesser von 1 bis 8 µm und bis zu einer Masse von 1,0 mg Cristobalit auf etwa 400 mg KBr anwendbar ist, unabhängig davon, welche der verwendeten Absorptionsbanden (621,5 cm-1 bzw., bei Abwesenheit von Quarz, 794,6 cm-1) zur quantitativen Auswertung eingesetzt wird. Bei Einsatz dieser sehr fein gemahlenen Cristobalite zur Gewinnung von feineren Fraktionen ist eine stärkere Verunreinigung durch amorphes SiO2 im Vergleich zum Quarz zu erwarten. Deswegen werden alle Cristobalit-Feinstaubproben (gewonnen z.B. durch Sedimentation oder Feinstaubsammlung mit dem MPG II) folgender mehrstufiger Reinigung unterzogen:
Durch die Natronlaugebehandlung ergeben sich hier zu vernachlässigende leichte Verschiebungen der Mediane der Korngrößenverteilung der Partikelgrößenfraktionen.
Zur Beschreibung des Gültigkeitsbereiches bei realen Proben wurden Soll-Faktorenf (analogF bei Quarz) errechnet. Der Soll-Faktor gibt an, ob der mittlere Partikeldurchmesser einer realen Probe im Bereich der Gültigkeit der Formel zur partikelgrößenkorrigierten Massenbestimmungmreal des Cristobalits liegt. In Tabelle 1 sind für die drei entwickelten Auswertungsmodelle die Werte des Achsenabschnittes B und der SteigungA, die Formeln zur Berechnung des Soll-Faktors sowie seine Gültigkeitsintervalle zusammengefasst.
Tabelle 1: Daten zur Berechnung des Cristobalit-Gehaltes und des Soll-Faktors f (Beispiel für das FTIR Bruker Vector 22)
| Auswertungsmodell | Ermittlung der Masse | Formel des Soll-Faktors |
Gültigkeitsintervall des Soll-Faktors |
| 680 - 585 cm-1 | A = 20,304 B = 0,686 |
f = (45,31 * H/F) - 1,53 | 0,77 - 1,21 |
| 630 - 610 cm-1 | A = 23,964 B = 1,320 |
f = (53,48 * H/F) - 2,95 | 0,78 - 1,22 |
| 900 - 707 cm-1 | A = 30,817 B = 0,271 |
f = (43,9 * H/F) - 0,386 | 0,65 - 1,41 |
Üblicherweise wird die von Quarz, Tridymit und amorphem SiO2 ungestörte Bande 621,5 cm-1 zur Auswertung herangezogen.
Bei Störungen im Flankenbereich dieser Bande kann eine alternative Auswertung in dem Auswertebereich 630 - 610 cm-1 benutzt werden, die Ermittlung der Höhen- und Flächenwerte erfolgt nach der Basislinienkorrektur (siehe Abbildung 7). Diese wird in der Regel durch Module der Auswertesoftware, die bei marktüblichen FTIR-Geräten mitgeliefert wird, ermöglicht. Die Durchführung einer Basislinienkorrektur erfordert Erfahrung. Sie muss vorab an Beispielspektren mit bekannten Störkomponenten gewonnen werden.
Abb. 7 Darstellung der Integration der basislinienkorrigierten Bande bei 621,5 cm-1 im Falle von Flankenstörungen
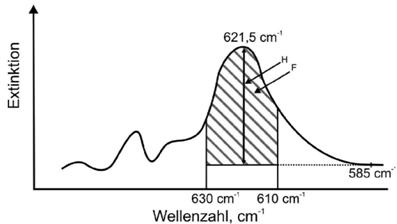
Die Grundlinie verläuft horizontal zur Wellenzahlenachse und wird durch den angegebenen Grundlinienpunkt auf der linken oder rechten Flanke der Absorptionsbande 621,5 cm-1 anhand empirischer Erfahrungen festgelegt (Hinweis: In Abbildung 7 ist die rechte Flanke mit dem fixen Grundlinienpunkt 585 cm-1 das Absorptionsminimum). Die Integrationsgrenzen sind festgelegt auf 630 und 610 cm-1, was etwa den Flankenpunkten der Halbwertsbreite der Absorptionsbande von Cristobalit entspricht. Die Anwendung dieses Auswertungsmodells ist nur unter der Voraussetzung möglich, dass eine der Flanken der Absorptionsbande von Cristobalit bzw. einer der festgelegten Basislinienstützpunkte 680 cm-1 oder 585 cm-1 störungsfrei ist.
Bei der Cristobalit-Analyse muss jedes Probenspektrum daraufhin überprüft werden, ob in den Basislinien an den Flanken der Bande des Cristobalits Überlagerungen mit Banden anderer Phasen auftreten. Liegen diese vor, kann versucht werden, mit einer Basislinienkorrektur unter Verwendung des gesamten Spektrums mit möglichst vielen Stützstellen Abhilfe zu schaffen. Wenn dies keinen Erfolg hat, ist eine Auswertung nicht möglich. In diesem Fall ist auf die röntgendiffraktometrische Analyse auszuweichen.
5 Beurteilung des Verfahrens
5.1 Präzision und Wiederfindung
Wiederfindungsrate: Die Wiederfindungsraten für Quarz und Cristobalit (Bande 621,5 cm-1) wurden für die verschiedenen Auswertungen differenziert ermittelt.
5.2 Bestimmungsgrenze
Die Bestimmungsgrenze wurde in Anlehnung an DIN EN 32.645 [ 8] für Quarz und Cristobalit anhand der Kalibrierfunktion im unteren Messbereich (siehe Abschnitt 3.1) und der Leerwertmethode ermittelt. Beide Verfahren liefern vergleichbare Ergebnisse.
Die Analyse reiner Quarz- bzw. Cristobalit-Stäube ergibt dabei absolute Bestimmungsgrenzen von 0,01 mg für Quarz und für Cristobalit (statistische Sicherheit 95 %, k = 3). Diese günstigen Werte werden jedoch unter realen Bedingungen bei der Analyse staubbeaufschlagter Filter zum Teil nicht erreicht. Die Bestimmungsgrenze liegt erfahrungsgemäß dann jeweils bei rund 0,03 mg absolut. Bezogen auf die verschiedenen Probenahmesysteme ergeben sich die in Tabelle 2 aufgeführten Werte der relativen Bestimmungsgrenze für Quarz und Cristobalit (Bande 621,5 cm-1).
Tabelle 2: Relative Bestimmungsgrenzen für Quarz und Cristobalit
| Probe- nahme- system |
Filterdurch- messer |
Luftvolu- menstrom |
Probe- nahmedauer |
Probeluft- volumen |
relative Bestimmungsgrenze |
|
| [mm] | [m3/h] | [h] | [m3] | ideal [mg/m3] |
real *) [mg/m3] |
|
| VC 25F | 150 | 22,5 | 2 8 |
45 180 |
0,0004 0,0001 |
0,0013 0,0003 |
| PM 4F | 70 | 4,0 | 2 8 |
8 32 |
0,001 0,0003 |
0,004 0,001 |
| FSP-10 | 37 | 0,6 | 2 8 |
1,2 4,8 |
0,008 0,002 |
0,025 0,006 |
| FSP-BIA | 37 | 0,12 | 2 8 |
0,24 0,96 |
0,042 0,010 |
0,13 0,031 |
| MPG II | 47 | 2,8 | 2 8 |
5,6 22,4 |
0,0018 0,0004 |
0,0054 0,0013 |
| *) Bestimmungsgrenze bei ungünstiger Proben matrix oder Störungen im Bereich der relevanten Absorptionsbanden | ||||||
5.3 Erweiterte Messunsicherheit
Die erweiterte Messunsicherheit U nach DIN EN 482 [ 11] bei Einsatz der Probenahmesysteme FSP-10 (10 l/min) und PM 4F (4 m3/h) wurde unter Zugrundelegung der analytischen Präzision (Verfahren nach Abschnitt 4.3) von 2,0 % bei 0,5 mg Quarz bzw. 1,9 % bei 0,43 mg Cristobalit für die ideale und reale absolute Bestimmungsgrenze (0,01 bzw. 0,03 mg) für Quarz und Cristobalit abgeschätzt [ 4, 7].
Bei Einsatz des Probenahmesystems PM4F beträgt die erweiterte Messunsicherheit für Quarz- bzw. Cristobalit-Konzentrationen von 0,01, 0,05, 0,10 und 0,15 mg/m3 bei einer Probenahmedauer zwischen 2 und 8 Stunden rund 24 %.
Für das Probenahmesystem FSP-10 ergibt sich eine erweiterte Messunsicherheit von 24 bis 26 %, mit Ausnahme der 2-stündigen Probenahme bei der Konzentration 0,01 mg/m3. Hier errechnet sich ein Wert von 30 bzw. 56 % (in Bezug auf die ideale bzw. die reale Bestimmungsgrenze) für die erweiterte Messunsicherheit.
Eine detaillierte Berechnung der erweiterten Messunsicherheit nach DIN EN 482 [ 11] findet sich beispielhaft in [ 7] und [ 12].
Bei der Abschätzung der Messunsicherheit bei sehr niedrigen Konzentrationen im Bereich von 0,01 mg/m3 sollte im Einzelfall auch der Einfluss der ubiquitären Belastung berücksichtigt werden. Zu bedenken ist, dass die ubiquitäre Belastung mit Quarz-A-Staub in der Luft in der Größenordnung von 0,001 mg/m3 liegt, durchaus aber auch Werte von bis zu etwa 0,01 mg/m3 erreichen kann.
5.4 Querempfindlichkeiten
Zur Identifikation von Querempfindlichkeiten durch andere Minerale bei der quantitativen Auswertung eines IR-Spektrums ist es zunächst empfehlenswert, eine qualitative Interpretation durchzuführen. So werden durch die Aufnahme des gesamten IR-Spektrums im Bereich der Wellenzahlen von 4000 cm-1 bis 350 cm-1 fast immer auch Hinweise auf die Existenz anderer Bestandteile neben Quarz bzw. Cristobalit erhalten. Spektren einiger möglicherweise störender Phasen sind im Anhang dargestellt. Darüber hinaus können zusätzliche Informationen, wie z.B. Angaben zur Herkunft der Probe (Arbeitsbereich), eine zielgerichtete Suche nach in der Probe enthaltenen Komponenten wesentlich erleichtern. Insgesamt gibt die qualitative Interpretation so Aufschluss darüber, wie mögliche Querempfindlichkeiten eliminiert oder berücksichtigt werden können.
Liegen Querempfindlichkeiten durch Carbonate, bestimmte Oxide und andere salzsäurelösliche Verbindungen vor, lassen sich diese wie folgt eliminieren:
Störungen der Auswertebande von Quarz
Für die Quarzanalyse mit Partikelgrößenkorrektur nach Abschnitt 4.3 wurden an synthetischen Mischproben aus Quarz-A-Staub und anderen Mineralen für die in Tabelle 3 zusammengestellten Mischungen Querempfindlichkeiten festgestellt, die für Quarz eine Abweichung der Wiederfindung von mehr als ± 10 % ergeben.
Die größten Störungen liegen bei Albit, Orthoklas, Mikroklin und Sillimanit vor. Cristobalit zeigt ab ca. 50 % Anteil in Mischung mit Quarz eine starke Querempfindlichkeit. Die genannten Störungen sind kaum durch eine gezielte Probenvorbehandlung zu eliminieren. Eine alternative Quantifizierung des Quarzgehaltes kann in Einzelfällen die Integration der Quarzbande bei 695 cm-1 liefern (geringer Partikelgrößeneinfluss) oder die Spektrensubtraktion gemäß Abschnitt 4.2; im Regelfall sollte ergänzend eine röntgendiffraktometrische Analyse erfolgen.
Bei folgenden Mischungen aus Quarz und anderen Mineralen blieb der Einfluss auf die Wiederfindung bei unter ± 10 %: Oligoklas, Bytownit, Kaolinit, Portlandzement, Montmorillonit, Labradorit, Talk, Schamotte, Fällungskieselsäure (amorph), Volclay (Natrium-Bentonit).
Tabelle 3: Relevante Querempfindlichkeiten bei der Bestimmung von Quarz im Bereich der Doppelbande bei 798 und 779 cm-1 gemäß dem Auswerteverfahren nach Abschnitt 4.3 durch andere Mineralphasen
| Mineral | Mineralanteil in Mischung [%] |
Wiederfindung Quarz [%] |
| Orthoklas | 17 38 50 |
110 137 150 |
| Mikroklin | 19 37 53 |
106 117 137 |
| Albit | 17 37 52 |
85 75 59 |
| Sillimanit | 21 38 49 |
80 59 44 |
| Cristobalit | 49 66 |
90 39 |
Störungen der Auswertebande von Cristobalit
Bedingt durch die vielen Querempfindlichkeiten ist das infrarotspektroskopische Verfahren der Cristobalit-Bestimmung eher für Arbeitsplätze geeignet, an denen keine die Cristobalit-IR-Analyse störenden Stoffe auftreten. Im Zweifelsfall sollte eine zusätzliche röntgendiffraktometrische Analyse, gegebenenfalls nach Rückgewinnung des Staubes durch Auflösung der KBr-tablette, erfolgen.
Querempfindlichkeiten der Cristobalit-Analyse mit Partikelgrößenkorrektur nach Abschnitt 4.3
In einer zusätzlichen Probenbehandlung mittels Salzsäure können Störungen durch bestimmte Minerale entweder fast oder ganz eliminiert werden (Vorgehensweise siehe Abschnitt 5.4). Dies trifft z.B. auf Carbonate (Calcit, Dolomit, Magnesit), Oxide (Hämatit), Silikate (Wollastonit, Nephelin) und Sulfate (Anhydrit, Baryt) zu.
Eine Auflistung potenziell störender Minerale gibt Tabelle 4 wieder.
Tabelle 4: In Stäuben vorkommende Minerale, die zu Querempfindlichkeiten bei der Cristobalit-Bestimmung führen können
| Bezeichnung | Störungen [cm-1] der Cristobalit-Bande bei | |
| 621 cm-1 | 794,6 cm-1 | |
| Quarz | - | 798; 779 |
| Amorphe Kieselsäure | - | ca. 820 - 720 |
| Hämatit | 552; 596 | - |
| Korund | 607; 642 | 797 |
| Albit | 646; 587; 534 | 787; 761; 744; 724 |
| Orthoklas | 583; 605; 649 | 772; 729 |
| Mikroklin | 585; 646 | 772; 729 |
| Sanidin | 545; 586; 638 | 780; 725 |
| Wollastonit | 681; 645; 567 | 965; 930; 903 |
| Sillimanit | 573; 635; 690 | 889; 818; 750 |
| Talk | 535; 670; 691 | - |
| Kaolin (geglüht bei 550 °C) | - | breite Bande 950 - 700 |
| Biotit | 680 | 730 |
| Phlogopit | 692 | 816; 732 |
| Anhydrit | 595; 613; 677 | - |
| Baryt | 610; 639 | - |
| Calcit | - | 876; 712 |
Vorgehensweise bei der Auswertung der Bande bei 621,5 cm-1
Korund, Orthoklas, Mikroklin, Sanidin und Sillimanit stören auch schon bei kleinen Gehalten; in der Regel müssen solche Proben röntgendiffraktometrisch analysiert werden.
Amorphe Kieselsäure und geglühter Kaolin (Metakaolinit) stören die Analyse nicht. Quarz stört nur, wenn eine hohe Quarzkonzentration neben einer niedrigen Cristobalit-Konzentration auftritt. Störungen durch Talk, Biotit und Phlogopit können durch Anwendung des Auswertungsmodells 630 - 610 cm-1 minimiert werden (siehe Abschnitt 4.3 zu Cristobalit). Anhydrit, Calcit und Baryt stören nach der in Abschnitt 5.4 angegebenen Salzsäurebehandlung nicht. Das Entfernen von Baryt erfordert aber bei größeren Anteilen im Staubgemisch größere Salzsäuremengen und eine längere Behandlungsdauer.
Störungen durch Wollastonit lassen sich durch Salzsäurevorbehandlung vermeiden (Nachteil: eventuell Bildung amorpher Kieselsäure).
Weitere Hinweise zur Minimierung von Störungen im Flankenbereich der Auswertebanden finden sich in [ 7].
6 Literatur
| [ 1] | Hebisch, R., Fricke, H.-H., Hahn, J.-U., Lahaniatis, M., Maschmeier, C.-P., Mattenklott, M. Probenahme und Bestimmung von Aerosolen und deren Inhaltsstoffen In: Deutsche Forschungsgemeinschaft (DFG): Luftanalysen; Analytische Methoden zur Prüfung gesundheitsschädlicher Arbeitsstoffe. Band 1, 14. Lieferung, 2005, 40 S. Wiley-VCH, Weinheim 2005 |
| [ 2] | DIN EN 481 Arbeitsplatzatmosphäre; Festlegung der Teilchengrößenverteilung zur Messung luftgetragener Partikel Beuth-Verlag, Berlin 1993 |
| [ 3] | Fricke, H.-H. Wissenschaftlicher Abschlussbericht: Untersuchungen zur Entwicklung eines analytischen Verfahrens zur Einzelbestimmung verschiedener Tonmineralien bei Anwesenheit von Quarz, Kohle und Stäuben aus Baustoffen unter besonderer Berücksichtigung kleiner Substanzmengen (Forschung Nr. 7260/03/048/01) Kommission der europäischen Gemeinschaften (Hrsg.), IGF Bochum, 1990 |
| [ 4] | Tribus, O. Infrarotspektroskopische Untersuchungen am Beispiel der quantitativen Bestimmung von Quarz und der qualitativen Bestimmung von Asbest Masterarbeit an der Fakultät für Chemie und Biochemie an der Ruhr-Universität Bochum, 2006 |
| [ 5] | Baumann, H. Oberflächeneigenschaften und Auflösungsverhalten von Siliziumdioxid In: Beiträge zur Silikose-Forschung, 85, (1965) 1 - 49 |
| [ 6] | Baumann, H., Rasche, B. Bericht des Silikose-Forschungsinstitutes der Bergbau-Berufsgenossenschaft, 6, (1971) 51 |
| [ 7] | Reinhold, O. Anwendung der FTIR-Spektroskopie für die quantitative Cristobalitanalyse im Arbeitsschutzbereich Dissertation an der Fakultät für Chemie und Biochemie an der Ruhr-Universität Bochum, 2009 Als Download unter: wwwbrs.ub.ruhrunibochum.de/netahtml/HSS/Diss/Reinhold Olga/diss.pdf |
| [ 8] | DIN EN 32645 Chemische Analytik - Nachweis-, Erfassungs- und Bestimmungsgrenze unter Wiederholbedingungen - Begriffe, Verfahren, Auswertung Beuth Verlag, Berlin 2008 |
| [ 9] | BGI 505-30 Verfahren zur Bestimmung der Massenanteile von Chrysotilasbest und Amphibolasbesten Carl Heymanns Verlag, Köln 1991 |
| [ 10] | Health and Safety Laboratory - HSE Crystalline silica in respirable airborne dusts - Directon-filter analyses by infrared spectroscopy and X-ray diffraction (MDHS 101). Methods for the Determination of Hazardous Substances (MDHS) HSE Books, Sudbury (2005) Als Download unter: www.hse.gov.uk |
| [ 11] | DIN EN 482 Arbeitsplatzatmosphäre - Allgemeine Anforderungen an die Leistungsfähigkeit von Verfahren zur Messung chemischer Arbeitsstoffe Beuth Verlag, Berlin 2006 |
| [ 12] | BGI/GUV-I 505-82 Verfahren zur Bestimmung von Quarz und Cristobalit - Verfahren 02: Röntgendiffraktometrie DGUV, in Vorbereitung |
| Spektren von Quarz, Cristobalit und typischen Störsubstanzen | Anhang 1 |
Für jede Substanz ist zum einen das gesamte IR-Spektrum dargestellt. Zum anderen ist zusätzlich der für die Auswertung von Quarz und Cristobalit relevante Bereich des Spektrums zwischen 550 und 950 cm-1 vergrößert dargestellt, um die Art der Querempfindlichkeit für die Quarz- bzw. Cristobalit-Bestimmung besser erkennbar zu machen.
Abb. A.1 FTI R-Spektrum von Quarz
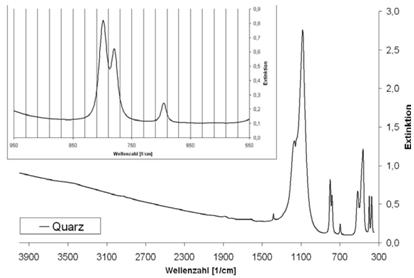
Abb. A.2 FTIR-Spektrum von Cristobalit
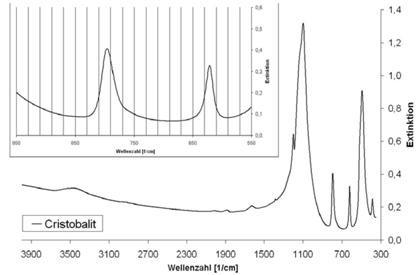
Abb. A.3 FTIR-Spektrum von Albit
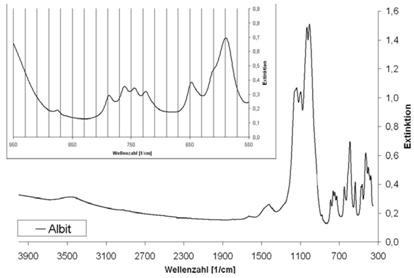
Abb. A.4 FTIR-Spektrum von amorpher Kieselsäure
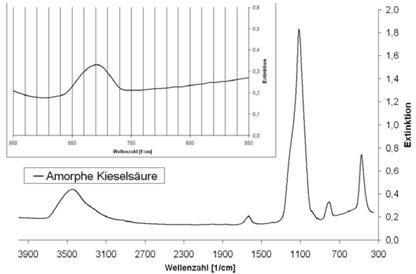
Abb. A.5 FTIR-Spektrum von Anhydrit
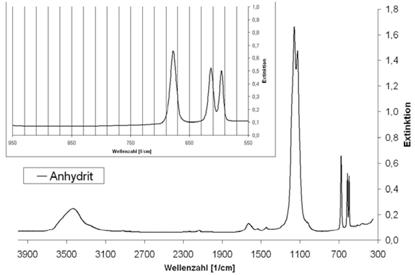
Abb. A.6 FTIR-Spektrum von Baryt
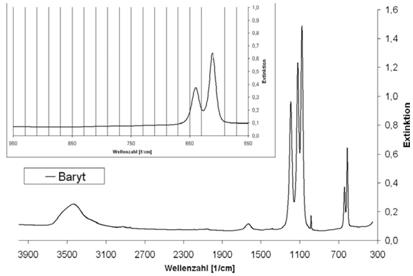
Abb. A.7 FTIR-Spektrum von Biotit
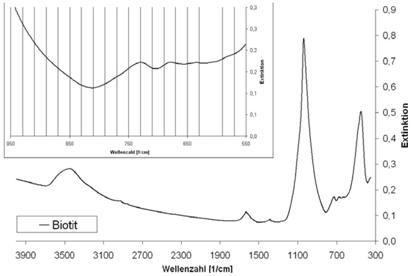
Abb. A.8 FTIR-Spektrum von Hämatit
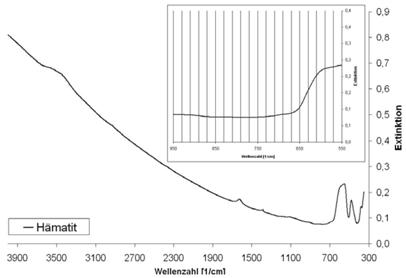
Abb. A.9 FTIR-Spektrum von geglühtem Kaolin

Abb. A.10 FTIR-Spektrum von Korund
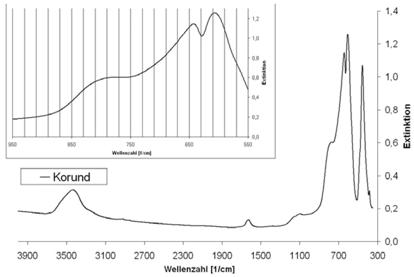
Abb. A.11 FTIR-Spektrum von Mikroklin

Abb. A.12 FTIR-Spektrum von Orthoklas
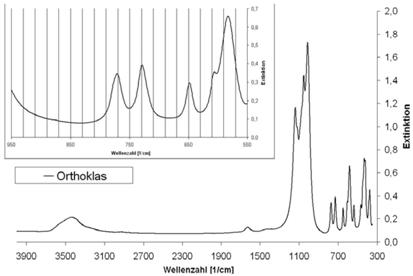
Abb. A.13 FTIR-Spektrum von Phlogopit
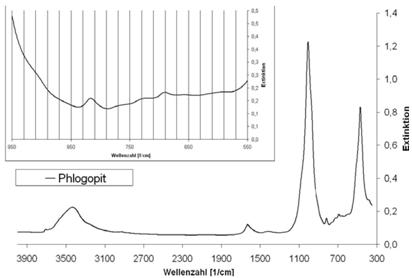
Abb. A.14 FTIR-Spektrum von Sanidin
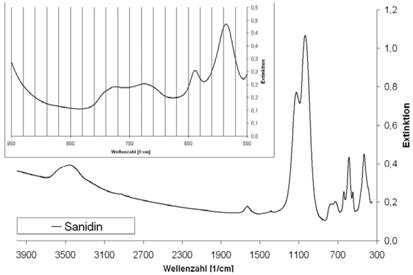
Abb. A.15 FTIR-Spektrum von Sillimanit
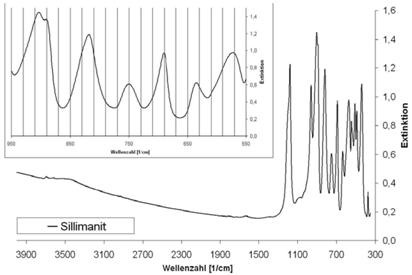
Abb. A.16 FTI R-Spektrum von Talk
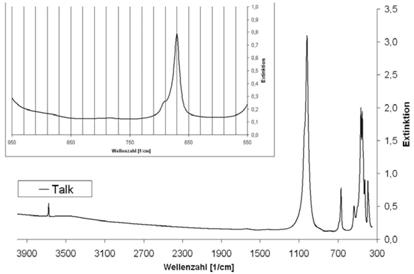
Abb. A.17 FTIR-Spektrum von Wollastonit

 |
ENDE | |
(Stand: 21.07.2025)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: ab 105.- € netto
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion