 Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk; BGI / DGUV-I

| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk; BGI / DGUV-I |
|
BGI 505-47 / DGUV Information 213-547 - Verfahren zur Bestimmung von 2,3,7,8-substituierten polychlorierten Dibenzodioxinen (PCDD) und Dibenzofuranen (PCDF)
Von den Berufsgenossenschaften anerkannte Analysenverfahren zur Feststellung der Konzentrationen krebserzeugender Arbeitsstoffe in der Luft in Arbeitsbereichen
Berufsgenossenschaftliche Informationen für Sicherheit und Gesundheit bei der Arbeit (BGI)
(bisherige ZH 1/120.47)
(Ausgabe 07/1997)
| Redaktioneller Hinweis: Berufsgenossenschaften sind gemäß § 210 SGB VII Behörden; ihre amtlichen Veröffentlichungen nach § 15 SGB VII unterliegen gemäß § 5 Abs. 2 UrhG keinem Urheberrechtsschutz. |
Zurückgezogen, nur zur Information
Erprobtes und von den Berufsgenossenschaften anerkanntes, diskontinuierliches Verfahren zur Bestimmung von PCDD und PCDF in Arbeitsbereichen.
Ortsfeste Probenahme von einatembarem Staub nach EN 481 für Messungen zur Beurteilung von Arbeitsbereichen.
Probenahme mit Pumpe, Filterabscheidung sowie Adsorption an Polyetherurethanschaum, Gaschromatographie nach extraktiver Desorption und Aufreinigung des Extraktes, hochauflösende massenselektive Detektion
"PCDD/PCDF-1 GC-HRMS".
Kurzfassung
Mit diesem Verfahren wird die über die Probenahmedauer gemittelte Konzentration 2,3,7,8-substituierter polychlorierter Dibenzodioxine und Dibenzofurane (PCDD/PCDF) im Arbeitsbereich bestimmt.
| Messprinzip: | Mit Hilfe einer Pumpe wird ein definiertes Luftvolumen durch ein integriertes Sammelsystem bestehend aus Probenahmesonde, Glasfaser-Tiefenfilter und nachgeschaltetem Polyetherurethanschaum auf TDI 1 -Basis (PUR-Schaum) gesaugt. Hierbei wird die einatembare Fraktion der partikelförmigen oder partikelgebundenen PCDD/PCDF angesaugt und auf dem Filter abgeschieden bzw. zusammen mit filtergängigen Anteilen am Schaum adsorbiert. Die Extraktion erfolgt durch Toluol im Soxhlet-Extraktor. Die extrahierten PCDD/PCDF werden von störenden Begleitstoffen durch kombinierte Aufbereitungsschritte abgetrennt und angereichert. Die gereinigten Extrakte werden gaschromatographisch aufgetrennt und die PCDD/PCDF durch hochauflösende Massenspektrometrie (HRMS 2) selektiv bestimmt. Probenahme, Probenaufbereitung und analytische Bestimmung werden durch mitgeführte isotopenmarkierte PCDD/PCDF-Standards (Isotopenverdünnungsmethode) kontrolliert. Die quantitative Auswertung erfolgt mittels dieser internen Standards. Die Angabe des Analysenergebnisses erfolgt in Toxizitätsäquivalenten (TE) unter Berücksichtigung von Toxizitätsäquivalent-Faktoren (TEF). |
| Technische Daten: | |
| Bestimmungsgrenzen (pro Kongener): | |
| absolut: | 0,3 pg TCDD/TCDF, PeCDD/PeCDF, HxCDD/HxCDF, 1 pg HpCDD/HpCDF und 3 pg OCDD/OCDF |
| relativ: | 0,15 pg/m3 an TCDD/TCDF, PeCDD/PeCDF, HxCDD/HxCDF, 0,5 pg/m3 an HpCDD/HpCDF und 1,5 pg/m3 an OCDD/OCDF bei 20 m3 Probeluft, 30 µl Probelösung und 3 µl Injektionsvolumen. |
| Selektivität: | Die Selektivität des Verfahrens hängt vor allem von der massenspektrometrischen Auflösung, von der Art der verwendeten Trennsäule und von Matrixbegleitstoffen ab. In der Praxis haben sich die angegebenen GC-Trennsäulen bewährt. |
| Vorteile: | Hohe Selektivität in Verbindung mit niedriger Bestimmungsgrenze. |
| Nachteile: | Keine personenbezogene Probenahme möglich, keine Anzeige von Konzentrationsspitzen; hoher probenahmetechnischer und analytischer Aufwand. |
| Apparativer Aufwand: | Probenahmeeinrichtung bestehend aus Probenahmekopf mit Probenahmesonde, Filterhalter und Adsorptionsrohr; Pumpe mit Gasmengenzähler oder Volumenstromanzeiger; GC/HRMS-Gerätekombination bestehend aus Gaschromatographen mit Kapillartrennsäule in direkter Kopplung mit hochauflösendem Massenspektrometer. |
Ausführliche Verfahrensbeschreibung
1 Vorbemerkungen
Die in diesem Verfahren beschriebene Vorgehensweise zur Bestimmung von PCDD/PCDF setzt wegen ihrer Thematik und Komplexität in besonderem Maße praktische Erfahrung in Probenahme- und Probenaufbereitungs-Techniken im Bereich der Ultra-Spurenanalytik/Gefahrstoffanalytik voraus. Ausführende Laboratorien müssen eine regelmäßige Qualitätskontrolle ausüben. Diese erfolgt beispielsweise intern durch Untersuchung von zertifizierten Standards und extern durch Teilnahme an Ringversuchen anerkannter Institutionen.
Dieses Verfahren ist durch sechs unabhängig voneinander arbeitende Laboratorien in einem Vergleichsversuch erarbeitet und validiert worden [1]. Die Laboratorien verwendeten alle das gleiche Probenahmesystem, aber unterschiedliche Verfahrensvarianten bei der Probenaufarbeitung und Analyse. Die Verfahrensvarianten des Vergleichsversuchs sind als Alternativen im Abschnitt 7 aufgeführt.
2 Probenahme
Die messtechnische Erfassung der partikelförmigen oder an Partikel gebundenen PCDD/PCDF erfolgt nach der Definition von einatembaren Staub entsprechend DIN/EN 481 [2] durch Abscheidung auf einem bindemittelfreien Glasfaser-Tiefenfilter. Die filtergängigen PCDD/PCDF-Anteile werden nachfolgend an PUR-Schaum adsorbiert.
Zur Differenzierung zwischen am Filter abgeschiedenen bzw. filtergängigen PCDD/PCDF können Filter und PUR-Schaum getrennt analysiert werden.
Die Verfahrenskenngrößen wurden mit dem unten beschriebenen Probenahmesystem ermittelt. Es eignet sich für stationäre Messungen, kann jedoch bei Bedarf auch im Arbeitsbereich bewegt werden, da es netzunabhängig betreibbar ist.
2.1 Geräte
Abb. 1: Probenahmeapparatur
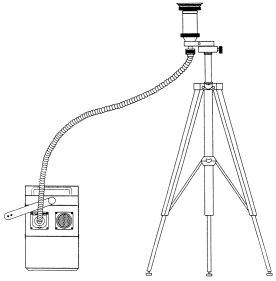
Die Bestandteile des Probenahmesystems sind nachfolgend aufgelistet:
Probenahmekopf PM4-G-D,
Ansaugpumpe PM4,
Stativ und Verbindungsschlauch zwischen Pumpe und Probenahmekopf,
Glasfaserfilter in einer Filterhalterung mit Deckeln,
PUR-Schaumpfropfen in einer 1-l-Braunglasflasche zur lichtgeschützten Aufbewahrung,
Transportkoffer für den Probenahmekopf,
Pinzette zur Schaumentnahme aus der Braunglasflasche,
Stativ und Druckschlauch zur Verbindung von Pumpe und Probenahmekopf,
Einweghandschuhe aus Polyethylen.
2.1.1 Probenahmekopf,
z.B. PM4-G-D nach BIA, Fa. Ströhlein.
Der Probenahmekopf selbst besteht im wesentlichen aus den Teilen Panoramakopfsonde und Probenträger, die zusammengeschraubt werden.
Abb. 2: Probenahmekopf
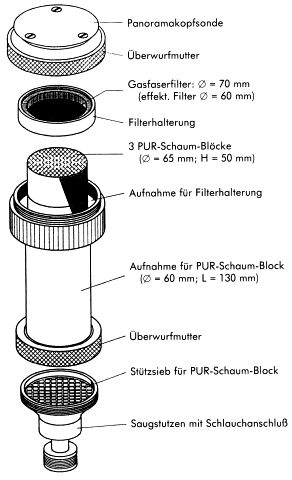
Die von der Probeluft berührten Teile sind aus V2A-Stahl gefertigt. Die Reinigung der Probenahmeköpfe vor jeder Probenahme erfolgt in drei Schritten:
Reinigung der Einzelteile in einer Spülmaschine mit handelsüblichem Spülmittel,
Ausspülen mit dest. Wasser,
Auswischen der Innenteile (gasberührten Teile) des Probenahmekopfes mit Aceton p.a.
Die Lagerung der Probenahmeköpfe erfolgt in dem gleichen dichtschließenden Aluminiumbehältern, die auch zum Transport benutzt werden.
2.1.2 Probenträger
Filter:
Bindemittelfreier Glasfasertiefenfilter, z.B. Typ MN 85/90 PF, Fa. Macherey-Nagel.
Zur Abscheidung der partikelförmig/partikelgebunden vorliegenden PCDD/PCDF wird ein Glasfaser-Tiefenfilter mit einem Durchmesser von 70 mm verwendet. Dieser Filter befindet sich so in einer Filterhalterung aus oberflächenmodifiziertem Aluminium, dass ein effektiver Filterdurchmesser von 60 mm verbleibt (s. Abb. 3). Zur Filterunterstützung wird ein Sieb von 0,4 mm Dicke und einer Maschenweite von 3,5 mm eingesetzt. Zum Transport der Filterkassetten werden oben und unten Deckel aus dem gleichen Material wie die Filterhalterung aufgelegt. Das System wird durch einen Spannbügel zusammengehalten.
PUR-Schaumpfropfen:
Polyetherurethanschaum auf TDI-Basis, z.B. Weichschaum der Fa. TPC, Klaus Ziemer GmbH.
Verwendet wird gereinigter Polyether-Weichschaum auf Basis von Toluylendiisocyanat mit einer Dichte von 20-25 kg/m3 und definierter Porosität. Die Porosität muß einem Gegendruck von 0,20 mbar äquivalent sein. Die einzelnen PUR-Schaumpfropfen haben einen Durchmesser von 65 mm und eine Höhe von 50 mm, wobei eine Überschreitung von 2 bis 3 mm zulässig ist. Die Reinigung der PUR-Schaumpfropfen erfolgt durch 24-stündige Soxhlet-Extraktion mit Toluol, 24-stündige Soxhlet-Extraktion mit Aceton und anschließendes Trocknen bei 40 °C im Vakuumtrockenschrank. Diese Arbeitsgänge sind unter Lichtausschluß durchzuführen. Drei PUR-Schaumpfropfen werden für eine Messung benötigt.
Anmerkung:
Polyesterschäume dürfen mangels Hydrolysestabilität nicht verwendet werden!
Abb. 3: Filterhalterung
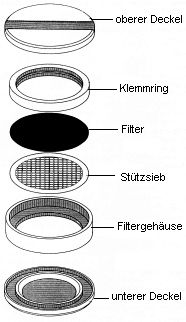
2.1.3 Ansaugpumpe,
z.B. Gravikon PM4, Fa. Ströhlein.
Das Gerät arbeitet mit einem pulsationsfreien geregelten Luftdurchsatz von 4 ± 0,1 m3/h unter Betriebsbedingungen. Der Luftdurchsatz wird abhängig von der Filterbelegung (Unterdruck max. 500 mm Wassersäule), dem Umgebungsdruck (zwischen 500 und 2000 hPa) und der Umgebungstemperatur (zwischen -10 °C und 50 °C) auf Konstanz geregelt.
Die Stromversorgung der Pumpe erfolgt wahlweise über Akkumulatoren oder Netzanschluß. Dadurch ist das Gerät für den Einsatz an mobilen Arbeitsplätzen geeignet, z.B. auf Fahrzeugen, in Krankabinen bzw. für Messungen an Arbeitsplätzen im Freien.
2.2 Chemikalien, Lösungen und Sorbentien
2.2.1 Lösemittel
Toluol p.a., zur Schaumvorreinigung.
Aceton p.a., zur Schaumvorreinigung und zur Reinigung des Probenahmekopfes.
2.2.2 Probenahmestandards,
z.B. Fa. Promochem, Fa. Cambridge Isotope Laboratories.
Für die leichterflüchtigen PCDD/PCDF:
1,2,3,4-13C12-TCDD, alternativ 1,2,3,4-13C6-TCDD, wenn keine Oleumbehandlung des Probeextraktes notwendig ist.
Optional für die leichterflüchtigen PCDD/PCDF:
2,3,7,8-37Cl4-TCDD, wenn Oleumbehandlung des Probeextraktes notwendig ist. ( Abschnitt 3.3.4) (kann bis zu 1 % natives 2,3,7,8-TCDD enthalten!)
Optional für die schwererflüchtigen PCDD/PCDF:
1,2,3,4,6,7,8-13C6-HpCDF.
2.2.3 Dotierlösung
Lösung von z.B. 1 ng/ml an 1,2,3,4-13C12-TCDD in Toluol
Herstellung:
10 µl einer käuflichen Standardlösung (Abschnitt 2.2.2) mit einem Gehalt von z.B. 1 µg/ml an 1,2,3,4-13C12-TCDD werden in 10 ml Toluol homogenisiert.
2.3 Durchführung
2.3.1 Aufbringen der Probenahmestandards
In die z.B. durch Schnittmarke gekennzeichnete Kopffläche eines der benötigten drei gereinigten PUR-Schaumpfropfen wird im Labor durch mehrmaliges punktförmiges Einstechen mittels einer Mikroliterspritze insgesamt 100 µl der Dotierlösung so injiziert, dass eine möglichst gleichmäßige Verteilung auf der Kopffläche erfolgt. Das Aufbringen erfolgt am einfachsten nach vorheriger Plazierung der Schäume im Probenahmekopf (Abschnitt 2.3.2), anderenfalls sind die zuvor dotierten Schaumpfropfen in einer Braunglasflasche getrennt von den nichtbehandelten zu lagern.
2.3.2 Aufbau der Probenahmeapparatur
Der erste Schritt zum Aufbau der Probenahmeapparatur ( Abb. 1) ist die Vorbereitung des Probenahmesystems, wobei mit dem PUR-Schaum begonnen wird. Die drei gereinigten PUR-Schaumpfropfen werden mittels der Pinzette aus dem Transportbehälter (Braunglasflaschen) in das an beiden Seiten zu öffnende Rohr plaziert. Da einer der drei PUR-Schaumpfropfen dotiert ist, ist auf die Reihenfolge des Einsatzes in den Probenahmekopf zu achten. Der dotierte Pfropfen ist dabei mit der dotierten Fläche zum Filter liegend direkt hinter diesem anzuordnen. Die Plazierung der PUR-Schaumpfropfen ist von entscheidender Bedeutung, damit ein guter Schluß zwischen Rohrinnenwand und Pfropfen besteht. Danach wird das Filter eingelegt und das Probenahmesystem zusammengeschraubt. Nach dem Anschließen der Schlauchverbindung zwischen Ansaugpumpe und Probenahmesystem und Aufstecken des Systems auf ein Stativ wird die Pumpe zur Probenahme in Betrieb gesetzt. Nach Abschluß der Messungen werden die Probenträger (Filter mit Filterhalter, PUR-Schaumpfropfen, Probenahmesystem) in die Transportbehälter verpackt zur Aufarbeitung umgehend in das Analysenlaboratorium gebracht.
3 Probenaufbereitung
Die aus Glasfaserfilter und drei PUR-Schaumpfropfen bestehende Probe wird im Soxhlet mit Toluol extrahiert. Die im Extrakt vorhandenen PCDD/PCDF werden von störenden Begleitstoffen durch Anwendung der nachfolgend beispielhaft und detailliert beschriebenen Aufreinigungsverfahren befreit.
Luftproben in Arbeitsbereichen enthalten im Gegensatz zu Proben aus dem Produkt- oder Emissionsbereich nur sehr wenig extrahierbare Matrix. Die Erfahrung zeigt, dass die in der angereicherten Luftprobe vorliegenden Matrixbestandteile häufig schon durch die Passage der Kombinationssäule bzw. durch Adsorptionschromatographie an Aluminiumoxid, wie in den Abschnitten 3.3.2 und 3.3.3 beschrieben, entfernt werden können. Aus diesem Grund sind weitere Aufreinigungsschritte nur in Ausnahmefällen notwendig. Wird durch die beiden Reinigungsschritte noch keine ausreichende Reinigung bewirkt, kann zusätzlich (neben einer möglichen Wiederholung der erstgenannten Aufreinigungsschritte) eine Schwefelsäure-Behandlung bzw. die Verwendung einer Aktivkohle/Celite-Einwegsäule, wie in den Abschnitten 3.3.4 und 3.3.5 beschrieben, erfolgreich sein.
Nachfolgend wird eines der erprobten und durch den Vergleichsversuch validierten Probenaufbereitungsschemata ausführlich beschrieben. Alternative Probenaufbereitungen sind in den Fließschemata des Abschnittes 7.4 aufgeführt.
3.1 Geräte
3.1.1 Probenextraktion
Eppendorfpipetten, im Volumenbereich von 100-500 µl,
Rundkolben mit Normschliff, 250 ml,
Soxhlet-Extraktor, 100 ml,
Rückflußkühler mit Schliffkern,
Heizhaube, 250 ml.
3.1.2 Extraktreinigung mit Kombinationssäule
Kunststoffsäule, 13 cm lang, i.D. 2,7 cm, mit Fritte und Auslauf, z.B. Fa. ICT Best.-Nr. A1213-1018, (alternativ kann eine Glassäule vergleichbarer Dimension benutzt werden)
Pasteurpipette,
Steilbrustflasche, 200 ml,
Erlenmeyerkolben mit Normschliff, 250 ml,
Braunglasflasche mit Normschliff, 250 ml,
Glasflasche mit Normschliff, 500 ml,
Rundkolben mit Normschliff, 100 ml,
Probengläschen mit Innenkonus, 1 ml.
3.1.3 Extraktreinigung mit Aluminiumoxid
Einweg-Kunststoffsäule mit Anschlußstück für Lösemittelreservoir, 14,5 cm lang, oberer Teil 2,5 cm lang, i.D. 1,7 cm bzw. unterer Teil 12 cm lang, i.D. 1,2 cm, z.B. Fa. Muromachi Kagaku Kogyo Kaisha LTD, Tokyo (Japan), (alternativ kann eine Glassäule vergleichbarer Dimension benutzt werden).
Pasteurpipette.
3.1.4 Extraktreinigung mit Schwefelsäure
Rundkolben mit Normschliff,
Rückflußkühler mit Schliffkern.
3.1.5 Extraktreinigung durch Säulenchromatographie an Aktivkohle/ Celite
Metallsäule, 12,5 cm lang, i. D. 0,4 cm (z.B. leere HPLC-Säule) oder Glassäule vergleichbarer Dimension, Schlauchpumpe zur Förderung des Eluenten.
3.2 Chemikalien und Lösungen
Die im folgenden aufgeführten Chemikalien sind von besonderer Reinheit hinsichtlich störender Chlorkohlenwasserstoffe.
3.2.1 Probenextraktion
Toluol, z.B. Fa. Promochem, Best.-Nr. 8092.
3.2.2 Extraktreinigung mit Kombinationssäule
Kieselgel, 70-230 mesh ASTM, z.B. Fa. Merck, Art.-Nr. 7734,
Natriumsulfat, wasserfrei, z.B. Fa. Riedel de Haën, Art.-Nr. 13464,
Silbernitrat, z.B. Fa. Baker, Art.-Nr. 1182,
Kaliumhydroxid, z.B. Fa. Reininghaus, Art.-Nr. R 222,
Schwefelsäure, z.B. Fa. Riedel de Haën, Art.-Nr. 30741,
Toluol, z.B. Fa. Promochem, Best.-Nr. 8092,
n-Hexan, z.B. Fa. Promochem, Best.-Nr. 4159,
Stickstoff, Reinheit 99,998.
3.2.3 Extraktreinigung mit Aluminiumoxid
Aluminiumoxid a Super I für die Dioxinanalytik, z.B. Fa. ICN, Best.-Nr. 404592,
n-Hexan, z.B. Fa. Promochem, Best.-Nr. 4159,
Dichlormethan, z.B. Fa. Promochem, Best.-Nr. 3023,
Diethylether, z.B. Fa. Promochem, Best.-Nr. 3434.
3.2.4 Extraktreinigung mit Schwefelsäure
Schwefelsäure, z.B. Fa. Riedel de Haën, Art.-Nr. 30741,
n-Hexan, z.B. Fa. Promochem, Best.-Nr. 4159.
3.2.5 Extraktreinigung durch Säulenchromatographie an Aktivkohle/Celite
Aktivkohle AX-21, z.B. Fa. Andersen, Adrian, Michigan (USA),
Celite 545, Fa. Bayer AG, Leverkusen,
Toluol, Fa. Promochem, Best.-Nr. 8092,
Dichlormethan, z.B. Fa. Promochem, Best.-Nr. 3023,
n-Hexan, z.B. Fa. Promochem, Best.-Nr. 4159.
3.2.6 Cleanup-Standard
Als Cleanup-Standard dient eine Mischung von 2,3,7,8-13C-markierten PCDD/PCDF, die jeweils mindestens ein PCDD/PCDF pro Chlorierungsgrad enthalten muß. Diese PCDD/PCDF-Kongeneren dürfen nicht an anderer Stelle des Analysenverfahrens verwendet werden, Lösung von z.B. jeweils 1 ng/ml an 2,3,7,8-13C-markierten PCDD/PCDF in Toluol.
Herstellung:
Je 10 µl der käuflichen Standardlösungen (Abschnitt 2.2.2) mit einem Gehalt von z.B. 1 µg/ml an 2,3,7,8-13C-markierten PCDD/PCDF werden gemeinsam in 10 ml Toluol homogenisiert.
3.2.7 Spritzen-Standard
Der Spritzen-Standard dient zur Bestimmung der Wiederfindungsraten der internen Standards. Als Spritzen-Standard werden z.B. 3 µl einer Lösung von 100 pg/µl 1,2,3,4-13C6-TCDD der Messlösung zugesetzt, sofern dieses nicht schon als Probenahmestandard eingesetzt wurde.
Herstellung:
3 µl einer käuflichen Standardlösung (Abschnitt 2.2.2) mit einem Gehalt von z.B. 1 µg/ml an 1.2.3.4.-13C6-TCDD werden in 10 ml Toluol homogenisiert.
3.3 Durchführung
3.3.1 Probenextraktion
Die Innenwandung des Probenahmekopfes wird zweimal mit ca. 25 ml Toluol ausgespült und die Spüllösung mit dem Extrakt des Glasfaserfilters und der PUR-Schaumpfropfen vereinigt.
Unmittelbar vor der Extraktion wird das Glasfaserfilter bzw. ein PUR-Schaumpfropf mit z.B. 300 µl Cleanup-Standard (Abschnitt 3.2.6) dotiert. Zur Extraktion wird der PUR-Schaum und das Glasfaserfilter in einen Soxhlet-Extraktor gegeben. Für die Extraktion wird ein Volumen von ca. 150 ml Toluol benötigt. Die Extraktionsdauer beträgt mindestens 7 Stunden. Der Extrakt wird zusammen mit der Spüllösung ca. 1 ml am Rotationsverdampfer eingeengt.
3.3.2 Extraktreinigung mit Kombinationssäule
Kombinationssäule aus schwefelsäure-, kaliumhydroxid- und silbernitrat-belegtem Kieselgel. Bei Bedarf größere Mengen sind entsprechende Vielfache der nachfolgend aufgeführten Mengen zu verwenden.
Kieselgel/Schwefelsäure:
56 g Kieselgel werden in eine Steilbrustflasche eingewogen und 44 g konzentrierte Schwefelsäure zugegeben. Man schüttelt bis ein einheitliches pulveriges Material entstanden ist.
Kieselgel/Silbernitrat:
50 g Kieselgel werden in einen 250 ml Schliff-Erlenmeyerkolben eingewogen und 5,5 g Silbernitrat gelöst in 21,5 g Wasser portionsweise unter Schütteln zugefügt. Die Mischung läßt man 30 min stehen. Das belegte Kieselgel wird unter Stickstoffgegenstrom in ein auf ca. 70 °C erwärmtes Glasrohr eingefüllt und die Temperatur schrittweise während etwa 5 Stunden auf 120 °C erhöht. Nach dem vollständigen Entweichen des Wassers erhitzt man weitere 15 Stunden auf 125 °C. Danach wird auf Raumtemperatur abgekühlt, die Stickstoffzufuhr unterbrochen und das belegte Kieselgel in eine dicht schließende Schliff-Flasche aus Braunglas gefüllt.
Kieselgel/Kaliumhydroxid (40 % KOH):
193 g KOH werden in 700 ml Methanol gelöst und die resultierende Lösung filtriert. Man gibt 300 g Kieselgel hinzu und rührt 90 min lang bei 55 °C. Das Lösemittel wird am Rotationsverdampfer abdestilliert bis ein feinpulvriges Material vorliegt. Das belegte Kieselgel wird in ein dichtschließendes Gefäß gefüllt.
In die Kunststoffsäule werden der Reihe nach folgende Reagenzien eingefüllt, wobei man durch Klopfen gegen die Säule ein möglichst gleichmäßig gepacktes blasenfreies Bett erzeugt:
1,5 g Kieselgel/10 % Silbernitrat
1,0 g Kieselgel
5,0 g Kieselgel/44 % Schwefelsäure
1,0 g Kieselgel
2,5 g Kieselgel/40 % KOH
1,0 g Kieselgel
2,0 g Natriumsulfat, wasserfrei
Nach Vorwaschen der Säule mit ca. 50 ml n-Hexan/Toluol (95:5 v/v) gibt man den aufkonzentrierten Probenextrakt auf die Säule. Das Probengefäß wird mit 3 mal 5 ml n-Hexan/Toluol (95:5 v/v) nachgespült und die einzelnen Anteile auch auf die Säule gegeben. Man eluiert mit weiteren 60 ml des gleichen Lösemittel-Gemisches und sammelt das Eluat in einem 100 ml Rundkolben. Das Säuleneluat wird mittels Rotationsverdampfer auf etwa 10 ml eingeengt, in einen Spitzkolben überführt und weiter eingeengt sowie nach weiterer Überführung in ein Probengläschen anschließend im Stickstoff-Strom auf ein Endvolumen von ca. 500 µl eingestellt.
3.3.3 Extraktreinigung mit Aluminiumoxid
Adsorptionschromatographie an Aluminiumoxid:
Das unter Abschnitt 3.2.3 beschriebene basische Aluminiumoxid der Aktivitätsstufe Super I ist ohne jegliche Vorbehandlung einsetzbar. Empfohlen wird die Lagerung und Handhabung unter Feuchtigkeitsausschluß.
In die Kunststoffsäule werden 10 g basisches Aluminiumoxid eingefüllt. Man überschichtet dann mit 1 g Natriumsulfat. Die in ca. 5 ml n-Hexan gelöste Probe ( 3.3.2) wird aufgegeben, wobei das Probegefäß dreimal mit 10 ml n-Hexan/Dichlormethan (99:1 v/v) nachgespült wird und die Spüllösung ebenfalls aufgetragen wird. Nach dem vollständigen Einsickern der Lösungen in das Trägermaterial eluiert man mit 150 ml n-Hexan/Dichlormethan (99:1 v/v). Dieses Eluat kann nach Abschluß der Analyse verworfen werden.
Die PCDD/PCDF-Fraktion wird mit 75 ml Dichlormethan/Diethylether (9:1 v/v) von der Säule eluiert.
3.3.4 Extraktreinigung mit Schwefelsäure
Sollten die unter den Abschnitten 3.3.2 und 3.3.3 angeführten Aufarbeitungs-Schritte nicht ausreichen, um störende Begleitkomponenten für die GC/HRMS-Bestimmung zu eliminieren, kann dieser Reinigungsschritt erfolgreich sein.
Die Schwefelsäurebehandlung kann direkt am Rohextrakt durchgeführt werden. Untersuchungen [4] zeigen, dass durch Schwefelsäurebehandlung bei erhöhter Temperatur der überwiegende Teil an organischer Matrix beseitigt werden kann.
Mit konzentrierter Schwefelsäure imprägniertes Kieselgel (Herstellung siehe Abschnitt 3.3.2) wird dabei zum Rohextrakt in Mengen bis etwa 50 g zugegeben. Das Lösemittel wird abgedampft und der trockene Rückstand bei 70 °C über 20 min behandelt. Alternativ kann das Schwefelsäure/Kieselgel-Gemisch zur Lösung in n-Hexan gegeben werden, wobei man die Mischung 20 min lang unter Rückfluß erhitzt. Nachfolgend wird das n-Hexan abdekantiert und der Rückstand zweimal mit n-Hexan nachgewaschen.
3.3.5 Extraktreinigung durch Säulenchromatographie an Aktivkohle/Celite
Sollten die unter den Abschnitten 3.3.2 und 3.3.3 angeführten Aufarbeitungs-Schritte nicht ausreichen, um störende Begleitkomponenten für die GC/HRMS-Bestimmung zu eliminieren, kann dieser Reinigungsschritt erfolgreich sein.
PCDD/PCDF werden selektiv an der Aktivkohle adsorbiert, wenn n-Hexan/Dichlormethan (1:1 v/v) als mobile Phase verwendet wird. Zur Erzielung höherer Durchflußraten wird der Aktivkohle Celite beigemischt. Die adsorbierten Verbindungen lassen sich anschließend mit Toluol eluieren, wobei die Säule in umgekehrter Flußrichtung betrieben wird. Die Aufreinigung an Aktivkohle kann nach der Kombinationssäule (Abschnitt 3.3.2) durchgeführt werden.
Zur Herstellung des Aktivkohle/Celite-Gemisches werden 82 g Celite und 18 g Aktivkohle in einen 500 ml Rundkolben eingewogen. Nach Durchmischung mit 500 ml Dichlormethan destilliert man das Lösemittel bei einer Wasserbadtemperatur von etwa 40 °C am Rotationsverdampfer unter Vakuum ab. Das Adsorbens wird nachfolgend bei 40 °C eine Stunde unter Vakuum getrocknet. Als stationäre Phase verwendet man ca. 0,5 g des Aktivkohle/Celite-Gemisches, wobei eine möglichst kompakte Packung in der Säule erzielt werden sollte. Die Säule wird nacheinander mit 100 ml Toluol/Dichlormethan (1:1 v/v) und 50 ml n-Hexan/Dichlormethan (1:1 v/v) gespült. Die Probelösung wird auf 50 ml mit n-Hexan/Dichlormethan (1:1 v/v) aufgefüllt und aufgetragen. Anschließend wird das Probegefäß mit weiteren 50 ml ausgespült und diese Lösung ebenfalls auf die Säule gegeben. Nicht adsorbierbare Probenanteile werden anschließend mit 250 ml n-Hexan/Dichlormethan (1:1 v/v) eluiert. Durch inverse Elution mit 300 ml Toluol (von 80 °C) läßt sich dann die Dioxin- und Furanfraktion eluieren. Alternativ kann die PCDD/PCDF-Fraktion entsprechend der Technik von Stalling [3] über eine Aktivkohle/Glasfaser-Säule gegeben werden. Hierzu werden 0,5 g einer Aktivkohle-Glasfaser-Mischung (1:9 w/w) in die Chromatographiesäule gefüllt und 5 bis 10 ml einer Probelösung in Cyclohexan/Dichlormethan (1:1 v/v) aufgegeben. Die Säule wird nacheinander mit 70 ml Cyclohexan/Dichlormethan (1:1 v/v) und 50 ml Dichlormethan/Methanol/Toluol (70:20:5 v/v/v) gespült. Danach dreht man sie um 180° und eluiert mit 50 ml Toluol in Gegenrichtung.
3.3.6 Messlösung
Die aufgefangenen Rohextrakte werden jeweils auf ein Endvolumen von z.B. ca. 200 µl im Stickstoffstrom eingeengt und mit 3 µl Spritzenstandard (Abschnitt 3.2.7) versetzt.
4 Analytische Bestimmung
4.1 Geräte
| Gaschromatograph: | Varian 3400 mit Kaltaufgabesystem Gerstel KAS 2 |
| Massenspektrometer: | Finnigan MAT 90 |
| Die im folgenden beschriebenen Arbeitsbedingungen wurden mit folgender Trennsäule ermittelt: | |
| Trennsäule: | DB Dioxin, polar, Fa. J&W Scientific Länge: 60m, Innendurchmesser: 0,25 mm, Filmdicke: 0,15 m. |
| Trägergas: | Helium, 180 kPa |
4.2 Arbeitsbedingungen
4.2.1 Gaschromatograph
Die Bestimmung der 2,3,7,8-substituierten PCDD/PCDF erfolgt auf einer polaren GC-Trennsäule des Typs DB-Dioxin. Die Trennung kann auch auf anderen polaren Kapillarsäulen wie z.B. Restek Rtx 2330, Supelco SP 2331 oder Chrompack CP Sil 88 erfolgen. Bei signifikanten Verlusten der hepta- und octachlorierten PCDD/PCDF oder bei einer schlechten Trennung von 2,3,7,8- und 2,3,4,8-TCDF ist die Trennsäule auszutauschen. Alternativ können die HpCDD/HpCDF und OCDD/OCDF auch auf einer Kapillarsäule mit unpolarer Trennphase bestimmt werden (z.B. J&W DB-5, HP Ultra 2). Die Trennleistung der GC-Säulen wird durch Injektion einer Testlösung mit PCDD/PCDF überprüft. Die bedarfsweise Bestimmung der Homologensummen-Parameter kann auch an einer unpolaren Trennsäule erfolgen (vergleiche Tab. 3 in Abschnitt 6.1.2).
| Kaltaufgabesystem-Temperaturprogramm: | ||
| Injektion bei: | Anfangstemperatur: | 60 °C |
| Anfangshaltezeit: | 90 s | |
| Lösemittelausblendung (Toluol) bei: | Heizrate 1: | 2 °C/s |
| Zwischentemp: | 80 °C | |
| Zwischenhaltezeit: | 90 s | |
| splitlose Aufgabe bei: | Heizrate 2: | 12 °C/s |
| Endtemperatur: | 300 °C | |
| Endhaltezeit: | 600 s | |
| Ofentemperaturprogramm: | Anfangstemperatur: | 100 °C |
| Anfangshaltezeit: | 3 min | |
| Heizrate 1: | 20 °C/min | |
| Zwischentemp: | 220 °C | |
| Heizrate 2: | 5 °C/min | |
| Endtemperatur: | 250 °C | |
| Endhaltezeit: | 90 min | |
| GC/HRMS-Interface-Temperatur: | 250 °C | |
4.2.2 Massenspektrometer
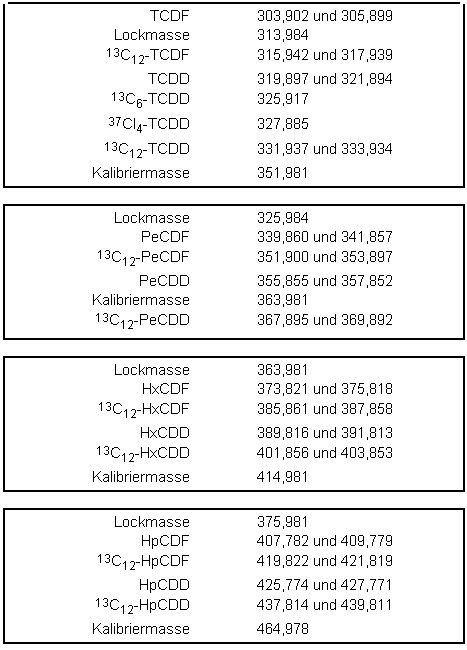
Das HRMS wird im SIM-Modus bei einer Auflösung von mindestens 5000 betrieben. Die Ionisation erfolgt unter Elektronenstoßbedingungen (70eV). Sowohl für die nativen als auch für die internen isotopenmarkierten Standards werden routinemäßig zwei ausgewählte Massenspuren registriert. Zur Absicherung können weitere Massenspuren aus dem Molekül-Ionencluster oder aus dem Cluster [M-COCl]+ herangezogen werden. Die Messzeit ist so zu wählen, dass jeder GC-Peak mit mindestens 10 Messpunkten abgebildet wird. Die entsprechenden Feinmassen zur selektiven Registrierung der PCDD/PCDF-Ionen sind in der folgenden Tabelle 1 enthalten.
Tabelle 1: Fein-Massen der Ionen zur Registrierung der PCDD/PCDF - (Kalibriersubstanz FC43)

4.3 Durchführung
4.3.1 Aufstellen der Kalibrierkurve
Die Kalibrierung erfolgt mit Lösungen, die die verwendeten isotopenmarkierten und zu bestimmenden nativen Standards enthalten (einschließlich Probenahme- und Spritzenstandards). Der Kalibrierbereich soll den erwarteten Konzentrationsbereich der Probe erfassen. Anhand der Kalibrierkurve werden die relativen Responsefaktoren der zu analysierenden Komponenten berechnet. Die Kalibrierhäufigkeit hängt von der Stabilität des GC/HRMS-Systems ab. Sie kann durch Injektion eines Kontrollkalibrierstandards überprüft werden.
Der Kalibrierfaktor wird für jedes zu bestimmende native PCDD/PCDF bezüglich eines festgelegten zugehörigen isotopenmarkierten PCDD/PCDF getrennt ermittelt.
Der Kalibrierfaktor fk des PCDD/PCDF-Kongeneren k bezüglich seines zugeordneten isotopenmarkierten Standards i wird nach Formel 1 berechnet: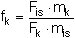 (1)
(1)
Es bedeuten:
| fk | = Kalibrierfaktor des PCDD/PCDF-Kongeneren k, |
| Fis | = Peakfläche des zugehörigen isotopenmarkierten Standards i aus dem Ionenmassen-Chromatogramm der Kalibrierlösung, |
| mk | = Masse des PCDD/PCDF-Kongeneren k in der Kalibrierlösung, |
| Fk | = Peakfläche des PCDD/PCDF-Kongeneren k aus dem Ionenmassen-Chromatogramm der Kalibrierlösung, |
| mis | = Masse des zugehörigen isotopenmarkierten Standards i in der Kalibrierlösung. |
Der Mittelwert des Kalibrierfaktors aus mehreren Analysen der Kalibrierlösungen ist für die Berechnung des Analysenergebnisses zu verwenden.
4.3.2 Identifizierung der PCDD/PCDF
Ein PCDD/PCDF ist identifiziert und bestimmbar, wenn folgende Kriterien erfüllt sind:
Das Signalverhältnis zwischen den beiden ausgewählten Isotopenpeaks entspricht dem theoretischen Wert innerhalb einer Bandbreite von 20 %.
Die Retentionszeitdifferenz zwischen nativem und isotopenmarkiertem PCDD/PCDF ist kürzer als 3 Sekunden. Bei der Bestimmung der Hepta- und OktaCDD/OktaCDF an polaren Säulen liegt die Toleranz bei 5 Sekunden. Das Signal-Rausch-Verhältnis beträgt mindestens 3:1 auf der intensitätsschwächeren der beiden Massenspuren.
4.3.3 Bestimmung der Wiederfindungsraten und der Blindwerte
Die Wiederfindungsraten der für die Probenahme und Probenaufbereitung zugesetzten internen Standards sollten jeweils über 0,5 liegen.
Bei der Anwendung des Verfahrens ist regelmäßig auf selektive Verluste einzelner Kongenere zu achten (siehe auch Abschnitt 6).
Über die gesamten Verfahrensschritte der Probenaufbereitung unter Einbeziehung der Probenahmeköpfe ist zusätzlich eine Blindwertkontrolle durchzuführen.
Um am Arbeitsplatz eine Konzentration von beispielsweise 50 pg TE/m3 an PCDD/PCDF zu kontrollieren, sollte der Summen-Blindwert nicht über 0,5 pg TE/m3 unter Einrechnung der Bestimmungsgrenzen liegen.
5 Berechnen des Analysenergebnisses
5.1 Analysenergebnis
Die Berechnung der Massenkonzentration eines einzelnen PCDD/PCDF-Kongeneren in der Probeluft erfolgt nach Formel 2: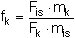 (2)
(2)
Es bedeuten:
| Fp | = Fläche des PCDD/PCDF-Kongeneren p aus dem Ionenmassen-Chromatogramm der Probelösung, |
| mqs | = Masse des isotopenmarkierten internen Standards q in der Probelösung in pg, |
| Fqs | = Fläche des isotopenmarkierten internen Standards q aus dem Ionenmassen-Chromatogramm der Probelösung, |
| fp | = mittlerer Responsefaktor des PCDD/PCDF-Kongeneren p nach Formel 1, bezogen auf den isotopenmarkierten Standard q, |
| cp | = Konzentration an PCDD/PCDF-Kongeneren p in der Probeluft in pg/m3, |
| V | = Probeluftvolumen in m3. |
Durch die Verwendung der Cleanup-Standards nach Abschnitt 3.3.1 vor der Extraktion ist die Wiederfindungsrate aus der Probenaufbereitung in den Ergebnissen berücksichtigt. Die Wiederfindungsraten der Probenahmestandards werden nur zur Information angegeben.
Aus den bekannten Kalibrierfaktoren des Spritzenstandards (bezogen auf die dotierten isotopenmarkierten PCDD/PCDF-Kongeneren) werden die individuellen Wiederfindungsraten dieser PCDD/PCDF-Kongeneren für die einzelnen Verfahrensschritte berechnet.
5.2 Toxizitätsäquivalente
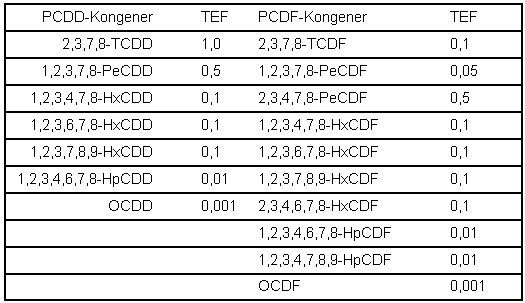
Die Berechnung des Gesamtgehalts an PCDD/PCDF in der Luft erfolgt nach Formel 3 unter Anwendung der Toxizitätsäquivalent-Faktoren (TEF) nach NATO/CCMS ( Tab. 2). Falls die Konzentration eines Kongeneren unterhalb der Bestimmungsgrenze liegt, wird dessen TE-Anteil unter Einrechnung der halben Bestimmungsgrenze [5] ermittelt.
Zur Berechnung der Toxizitätsäquivalent-Konzentration CTE in pg/m3 wird zuerst die jeweilige ermittelte PCDD/PCDF-Kongener-Konzentration Cn mit dem zugehörigen TE-Faktor TEFn multipliziert. Sodann werden diese Produkte für alle 2,3,7,8-substituierten PCDD/PCDF-Kongeneren der Tabelle 2 (m = 17) aufaddiert.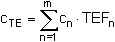 (3)
(3)
Es bedeuten:
| cn | = Massenkonzentration an PCDD/PCDF-Kongener n in der Probeluft in pg/m3, |
| cTE | = Toxizitätsäquivalent-Konzentration in der Probeluft in pg TE/m3, |
| TEFn | = TE-Faktor des PCDD/PCDF-Kongeneren n, |
| m | = 17, Gesamtzahl der (mit TEF versehenen) PCDD/PCDF-Kongeneren. |
Tabelle 2: Toxizitätsäquivalent-Faktoren (TEF) nach NATO/CCMS (1988) [7]
5.3 Ergebnisdarstellung
Die Angabe der kongenerenspezifischen Konzentration an PCDD/PCDF in der Luft erfolgt in pg/m3 nach Formel 2 auf zwei signifikante Stellen gerundet. Die Angabe des PCDD/PCDF-Gehalts der Luft erfolgt als Toxizitätsäquivalent-Konzentration in pg TE/m3 nach Formel 3 auf maximal zwei signifikante Stellen gerundet.
6 Beurteilung des Verfahrens
Zur Ermittlung der relativen Messunsicherheit des beschriebenen Verfahrens wurde ein Vergleichsversuch durchgeführt, bei dem sechs Laboratorien beteiligt waren. Hierzu wurden sechs Luftprobenahmen zeitgleich durchgeführt. Das Probeluftvolumen betrug 32 m3 bei einer Probenahmedauer von insgesamt acht Stunden. Die Probenahme erfolgte in der Halle eines Aluminium-Recycling-Betriebes, in der eine homogene Verteilung der PCDD/PCDF vorausgesetzt werden konnte. Die Filterproben wurden nach der Probenahme klimatisiert und dann zurückgewogen, um die jeweilige Staubmasse zu ermitteln. Anschließend wurden sie zusammen mit den zugehörigen drei PUR-Schaumpfropfen an die teilnehmenden Laboratorien versandt.
Die Bearbeitung der Proben erfolgte je nach Labor zu unterschiedlichen Zeitpunkten innerhalb von zwölf Wochen nach der Probenahme. Jedes Labor erhielt einen nicht beaufschlagten Satz an PUR-Schäumen mit einem Filter, um die Blindwerte der Probenträger und des gesamten Analysenverfahrens zu bestimmen. Zur Ermittlung der Messwerte bzw. Blindwerte wurden jeweils das Filter mit den drei PUR-Schaumpfropfen zusammen aufgearbeitet und analysiert. Die ausführliche Darstellung ist publiziert [1].
Die Ergebnisse des Vergleichsversuchs zeigen, dass das Verfahren geeignet ist, eine Konzentration von z.B. 50 pg TE/m3 an PCDD/PCDF in der Luft am Arbeitsplatz zu überwachen.
6.1 Genauigkeit und Wiederfindungsrate
6.1.1 Staubkonzentration
Die Konzentration des einatembaren Staubes lag im Mittel bei 0,24 mg/m3. Die relative Standardabweichung betrug 1,2 %. Diese Übereinstimmung der Staubwerte zeigt, dass bei der Probenahme keine systematischen Fehler in bezug auf den Standort einzelner Probenahmesysteme gemacht wurden.
6.1.2 PCDD/PCDF-Kongenere
Die von den einzelnen beteiligten Laboratorien ermittelten PCDD/PCDF-Konzentrationen gibt Tabelle 3 wieder.
Tabelle 3: PCDF-Konzentrationen und Standardabweichungen aus dem Vergleichsversuch [1]
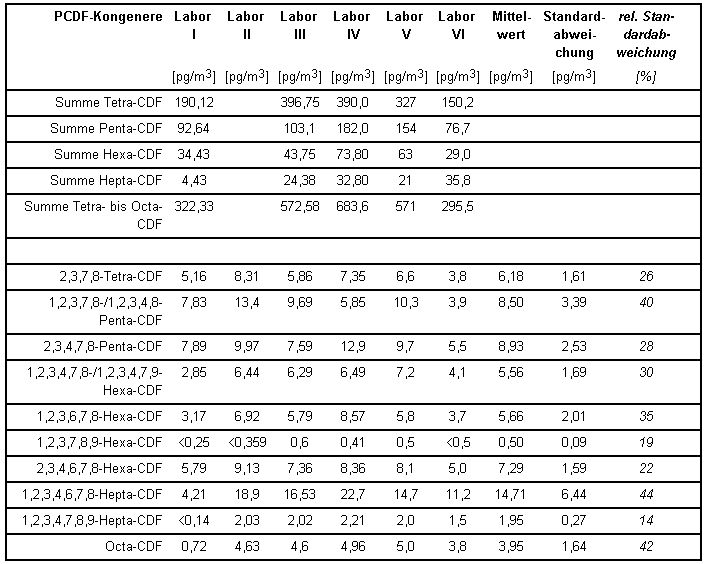
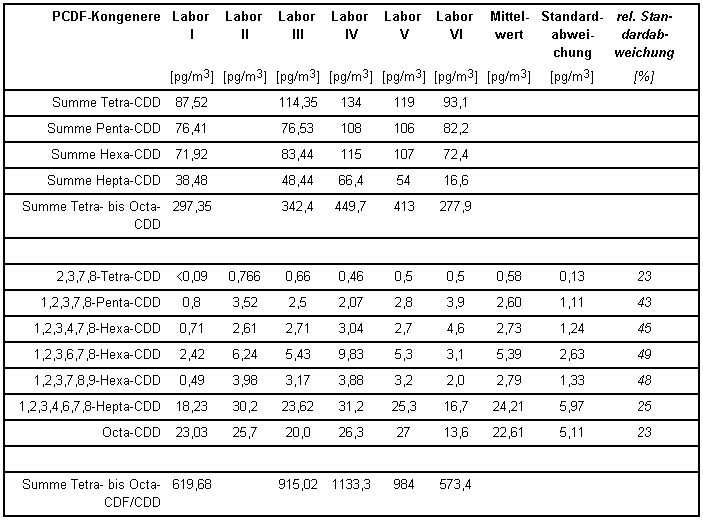
In der folgenden Tabelle 4 sind diese Konzentrationsangaben auf Toxizitäts-Äquivalent-Konzentrationen nach NATO/CCMS [7] umgerechnet und letztere aufsummiert als Endergebnis wiedergegeben. Die Werte beziehen sich jeweils auf die Summe aus partikelförmigen oder partikelgebundenen und filtergängigen PCDD/PCDF-Anteilen.
Aus der getrennten Auswertung von Filter und PUR-Schaum eines der teilnehmenden Labors ergab sich, dass ca. 30 % der Gesamtmasse PCDD/PCDF(TE) als filtergängiger Anteil am untersuchten Arbeitsplatz vorlag. Dies wird durch Ergebnisse aus Metallrecycling-Betrieben [6] bestätigt. In einer weiteren Spalte sind Ähnlichkeitsparameter der Kongenerenprofile angeführt. Nähere Erläuterungen und die Berechnung sind in Abschnitt 6.1.3 dargestellt.
Tabelle 4: TE-Konzentrationen der PCDD/PCDF und Ähnlichkeits-Parameter der Kongeneren-Profile (Abschnitt 6.1.3) aus dem Vergleichsversuch zur Verfahrensvalidierung
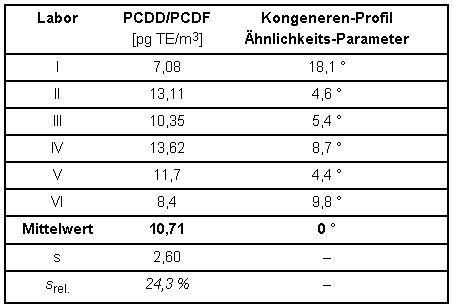
Aus den Ergebnissen errechnet sich als Messunsicherheit eine relative Standardabweichung für das Gesamtverfahren von ca. 25 %. Auch die Messunsicherheiten der Konzentrationsmittelwerte aus Tabelle 3 für die 17 bestimmten 2,3,7,8-substituierten Kongenere liegen noch unter 50 %, viele sogar im Bereich der Streuung der oben genannten TE-Konzentrationen aus Tabelle 4.
6.1.3 Kongeneren-Profil
Die Messergebnisse des Vergleichsversuchs wurden hinsichtlich der Übereinstimmung im Kongeneren-Profil mit einem mathematischen Verfahren überprüft, weil die auf TE-Einheiten reduzierten Messwerte sich zufällig ergeben könnten. Unter qualitativen Gesichtspunkten sollte die Kongenerenverteilung bei zeitgleicher Probenahme identisch sein. Erkennbare Unterschiede in der Konzentrationsverteilung der PCDD/PCDF-Kongeneren können auf selektive Verluste einzelner Kongeneren hinweisen.
Die Ähnlichkeit von Kongenerenverteilungen kann rechnerisch durch Interpretation der Messwerte eines Kongeneren-Kollektivs als Vektor (z.B. Messwerte der 17 PCDD/PCDF-Kongeneren einer Labor-Spalte in Tab. 3) durchgeführt werden [10]. Als Ähnlichkeitsparameter wird der Schnittwinkel zwischen zwei dieser Vektoren definiert, der nach Formel 4 über die Skalarprodukte berechnet wird: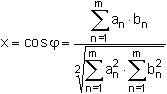 (4)
(4)
Es bedeuten:
| an | = Messwert des PCDD/PCDF-Kongeneren der Zeile n in der Spalte a der Tab. 5, |
| bn | = Messwert des PCDD/PCDF-Kongeneren der Zeile n in der Spalte b der Tab. 5, |
| x | = Cosinusbetrag des Winkel zwischen den als Richtungsvektoren betrachteten Messwerte-Spalten, |
| φ | = Winkelbetrag zwischen den als Richtungsvektoren betrachteten Messwerte-Spalten, |
| n | = Laufindex der Zeilen in der Tabelle 5, |
| m | = Gesamtzahl der zum Ähnlichkeitsvergleich herangezogenen Kongeneren einer Labor-Spalte in Tab. 5 (m = 17). |
Es handelt sich bei diesem Vorgehen um ein mathematisches Verfahren der Vektortransformation. Volle Ähnlichkeit (Übereinstimmung, Abhängigkeit) entspricht dem Schnittwinkel 0°, völlige Bezugslosigkeit (Unabhängigkeit, Orthogonalität) dem Schnittwinkel 90°. Der Vorteil der mathematischen Ähnlichkeitsbetrachtung liegt darin, dass die Ergebnisse Maßzahlen liefern, die zudem unabhängig von Berechnungsfaktoren (z.B. TE-Faktoren) sind, solange diese jeweils die gleichen PCDD/PCDF-Kongeneren betreffen.
Zur Verdeutlichung sind in folgender Tabelle 5 anhand zweier Spalten aus Tabelle 3 des Abschnitts 6.1.2, beispielhaft die Ähnlichkeits-Parameter zwischen den Kongeneren-Profilen (hier: Laboratorien V und VI) mit gerundeten Messwerten ermittelt. Hierbei sind die Werte unterhalb der analytischen Bestimmungsgrenze mit Null angesetzt worden, da sie bei diesem Vorgehen vernachlässigbar in das Ergebnis eingehen.
Tabelle 5: Berechnung der Ähnlichkeits-Parameter
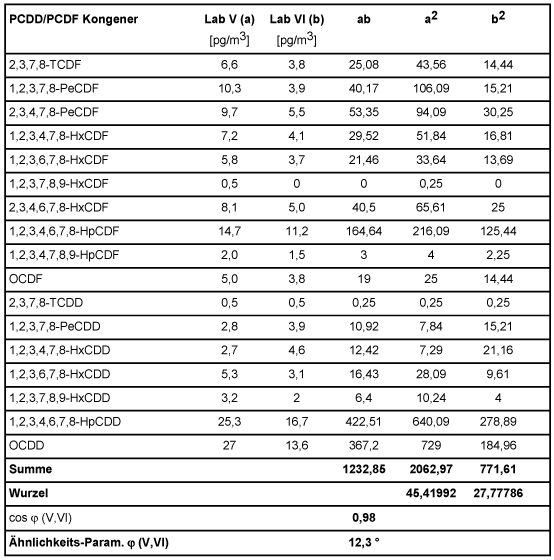
Die folgende Tabelle 6 enthält die auf beschriebene Weise ermittelten Schnittwinkel zwischen den Vektoren der Laboratorien der Tab. 3 untereinander, sowie mit dem Vektor der Mittelwerte
Tabelle 6: Ähnlichkeitsmatrix der Kongeneren-Profile Ähnlichkeitsparameter der Kongenerenprofil-Vektoren
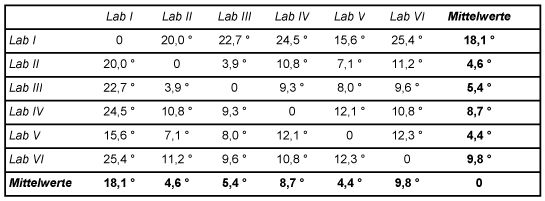
Die Schnittwinkel der Vektoren der beteiligten Laboratorien mit dem Vektor der Mittelwerte (Zeile bzw. Spalte "Mittelwerte" in obiger Tabelle) sind in folgender Abb. 4 graphisch dargestellt. Der Mittelwertvektor ist hierbei die horizontale Linie.
Abb. 4: Ähnlichkeit zwischen den Kongeneren-Profilen, Schnittwinkel
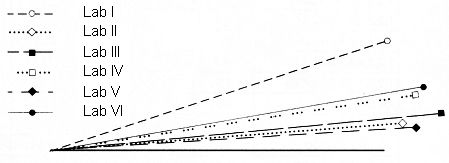
Die Ähnlichkeit der Kongeneren-Profile liegt innerhalb eines Bereichs von 4,4° bis 18,1° gegenüber dem Vektor der Mittelwerte. Dies ist ein Bereich der noch befriedigende Übereinstimmung darstellt.
6.2 Wiederfindungsraten
Die Wiederfindungsraten des Gesamtverfahrens können für alle Kongeneren innerhalb einer homologen Reihe als konstant angenommen werden.
Sie werden durch das Auswerteverfahren bedingt in der Ergebnisberechnung berücksichtigt ohne explizit bestimmt zu werden. Die Wiederfindungsrate des Probenahmestandards wurde separat ermittelt und lag mit einer Ausnahme oberhalb 0,7 [1].
6.3 Bestimmungsgrenzen
6.3.1 Leerwerte
Geeignete Verfahren sollen in der Regel die Überwachung eines Zehntels des jeweiligen Grenzwertes erlauben. Bei einem Luftgrenzwert von 50 pg TE/m3 entspricht dies einer PCDD/PCDF Konzentration von 5 pg TE/m3. Die Ergebnisse der Leerwertanalysen sind in Tabelle 7 enthalten. Sie zeigen, dass mit dem beschriebenen Verfahren bei achtstündiger Probenahme aus 32 m3 Probeluft genügend PCDD/PCDF angereichert werden konnten, um den notwendigen Abstand zwischen Grenzwert [9] und Leerwert für Arbeitsplatzmessungen zu erhalten.
Tabelle 7: TE-Leerwert-Konzentrationen an PCDD/PCDF aus dem Vergleichsversuch bezogen auf 32 m3 Probeluftvolumen.
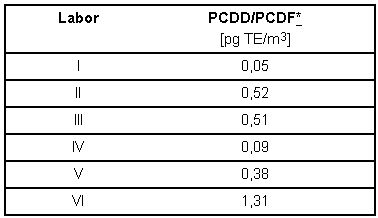
* Für nicht nachgewiesene Kongenere wurden die entsprechenden Bestimmungsgrenzen voll eingerechnet.
6.3.2 Bestimmungsgrenzen der PCDD/PCDF-Kongeneren
Unter optimalen Bedingungen ermöglicht das chromatographische Trennsystem in Verbindung mit einem hochauflösenden Massenspektrometer bei einer Auflösung von R = 10.000 folgende absolute Bestimmungsgrenzen:
0,3 pg TCDD/TCDF; PeCDD/PeCDF, HxCDD/HxCDF,
1 pg HpCDD/HpCDF und
3 pg OCDD bzw. OCDF
Für 30 µl Probelösung, 3 µl Injektionsvolumen und 20 m3 Probeluftvolumen ergeben sich hiermit folgende relative Bestimmungsgrenzen:
0,15 pg/m3 an TCDD/TCDF, PeCDD/PeCDF, HxCDD/HxCDF
0,5 pg/m3 an HpCDD/HpCDF und
1,5 pg/m3 an OCDD bzw. OCDF
6.4 Selektivität
Die Selektivität des Verfahrens hängt vor allem von der massenspektrometrischen Auflösung, von der Art der verwendeten Trennsäule und von Matrixbegleitstoffen ab. In der Praxis haben sich die angegebenen GC-Trennsäulen bewährt.
7 Bemerkungen
7.1 Qualitätssicherung
Leerwerte sind regelmäßig zu überprüfen und die ermittelten Werte zu protokollieren. Bei vergleichsweise hohen gemessenen Konzentrationen genügt es, dass der Leerwert zumindest um den Faktor 10 unter dem niedrigsten gemessenen Wert einer Probenserie liegt.
Die Überprüfung von Einzelschritten des Verfahrens (insbesondere Extraktion, Extraktreinigungsschritte und GC/HRMS-Messung, sowie Referenzstandards) kann durch regelmäßige Untersuchung von zertifizierten Referenz- oder Kontrollmaterialien und Standardlösungen erfolgen. Hierbei sollte die Wiederfindungsrate des überprüfbaren Verfahrensteils insgesamt nicht unter 0,5 liegen.
7.2 Lagerfähigkeit
Aus dem Vergleichsversuch leitet sich eine überprüfte Lagerfähigkeit der Proben von mindestens 12 Wochen ab.
7.3 Parallelbestimmung weiterer Stoffe
Das beschriebene Verfahren kann auch als Grundlage zur Bestimmung von polybromierten Dibenzodioxinen und polybromierten Dibenzofuranen dienen. Über Verfahrenskenngrößen, insbesondere die Probenaufbereitung, Kalibrierung und Wiederfindungsraten liegen keine vergleichbaren Untersuchungsergebnisse vor. Sie sind individuell zu erarbeiten.
7.4 Alternative Probenaufbereitungsverfahren
Die angewendeten Aufbereitungsschemata der einzelnen am Vergleichsversuch beteiligten Laboratorien bieten Alternativen und Ergänzungen zum beispielhaft beschriebenen Verfahren.
7.5 Alternative Arbeitsbedingungen für die analytische Bestimmung
Die tabellarisch aufgeführten Gerätebedingungen der einzelnen am Vergleichsversuch beteiligten Laboratorien bieten Alternativen zum beispielhaft beschriebenen Verfahren.
8 Hersteller
Herstellerangaben zum Abschnitt 2, Probenahme
Probenahmekopf Gravikon PM 4 GD und Probenahmepumpe Gravikon PM 4,
Fa. Ströhlein GmbH & Co.
Labor-, Mess- und Umwelttechnik
Girmeskreuzstr. 55
41564 Kaarst
Bindemittelfreie Glasfasertiefenfilter Typ MN 85/90 PF,
Fa. Macherey-Nagel
Postfach 101352
52313 Düren
Polyether-Weichschaum auf TDI-Basis, Dichte: 20 - 25 kg/m3,
Fa. TPC, Klaus Ziemer GmbH
Pommernstr. 96
68309 Mannheim
Herstellerangaben zum Abschnitt 3, Probenaufbereitung
Fa. Promochem
Postfach 101340
46469 Wesel
Fa. Cambridge Isotope Laboratories,
Mass. 01810-5413 U.S.A.
50 Frontage Road, Andover
Fa. Merck KGaA
Frankfurter Str. 250
64271 Darmstadt
Fa. ICT Handelsgesellschaft GmbH
Norsk-Data-Str. 3
61352 Bad Homburg
Fa. Riedel de Haën AG
30926 Seelze
Fa. Baker
Im Leuschenpark 4
64347 Griesheim
Fa. ICN-Biomedicals GmbH
Mühlengrabenstr. 10
53090 Meckenheim
Fa. Andersen
1415 E. Michiganstreet
Adrian, Michigan
49221 - 3499 (USA)
Fa. Reininghaus-Chemie
Joachimstr. 122
45309 Essen
Fa. Muromachi Kagaku Kogyo Kaisha LTD
Tokyo (Japan)
Herstellerangaben zum Abschnitt 4, Analytische Bestimmung
Gerstel Kaltaufgabesystem KAS 2,
Gerstel GmbH
Aktienstr. 232-234
45473 Mühlheim/Ruhr
Massenspektrometer MAT 90 oder MAT 95,
Fa. Finnigan MAT GmbH
Barkhausenstr. 2
28197 Bremen
Trennsäule DB Dioxin,
J & W Scientific Products GmbH
Horbeller Str. 15
50858 Köln
9 Literatur
| [ 1] | N. Lichtenstein; R. Stockmann, B. Bogdoll, J. Hosseinpour, K. Jaeger, H. Kieper, H. Linde, K. Schramm, Gefahrstoffe-Reinhaltung der Luft 56, 239 (1996), "Ringversuch zur Messung polychlorierter Dibenzodioxine und -furane an Arbeitsplätzen". |
| [ 2] | DIN EN 481, Beuth Verlag, Berlin, "Festlegung der Teilchengrößenverteilung zur Messung luftgetragener Partikel". |
| [ 3] | L.M. Schmith, D.L. Stalling, J.L. Johnson, Anal. Chem. 56, 1830 (1984), "Determination of part-per-trillion levels of polychlorinated dibenzofurans and dioxins in environmental samples". |
| [ 4] | J. Höckel, L. Düsterhöft, W. Körner, H. Hagenmaier Organohalogen Compounds Vol. 23, 135 (1995), "Modified Clean-Up Procedure for PCDD/PCDF under Toxicological, Ecological and Economical Aspects". |
| [ 5] | Anhang zur TRGS 102, Nr. 42, Carl Heymanns Verlag, Köln, "Technische Richtkonzentrationen (TRK) für gefährliche Stoffe". |
| [ 6] | J. Stockmann, U. Hahn, N. Lichtenstein, H.-D. Neumann, W. Schick, Staub-Reinhaltung der Luft 53, 389 (1993), "Polychlorierte Dibenzodioxine und -furane an Arbeitsplätzen". |
| [ 7] | NATO/Committee for the challenges of modern society, project for international information exchange on dioxins, furans and related chemicals CCMS Report No. 176, August 1988, "Toxicity equivalent factor method of risk assessment for complex mixtures of dioxins and related compounds". |
| [ 8] | DIN EN 482, Beuth Verlag, Berlin, "Allgemeine Anforderungen an Verfahren für Messung von chemischen Arbeitsstoffen". |
| [ 9] | TRGS 402, Carl Heymanns Verlag, Köln, "Ermittlung und Beurteilung der Konzentration gefährlicher Stoffe in der Luft in Arbeitsbereichen". |
| [ 10] | Alan Jeffrey, Mathematik für Naturwissenschaftler und Ingenieure, Band 1, Verlag Chemie (1973), "Vektoren, Differential- und Integralrechnung" (siehe auch DIN 38407-3). |
| [ 11] | E. Stahl, W. Schild, Pharmazeutische Biologie Bd. 4, 24 (1981) Gustav Fischer Verlag, Stuttgart, New York, "Drogenanalyse II". |
| zu 7.4 | Anlage |
Alternative Probenaufbereitungsverfahren
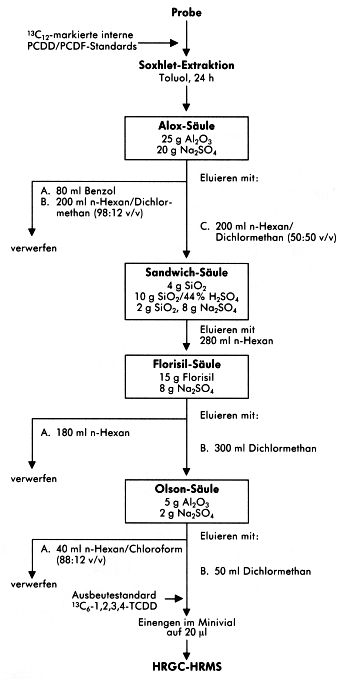
Abbildung 5
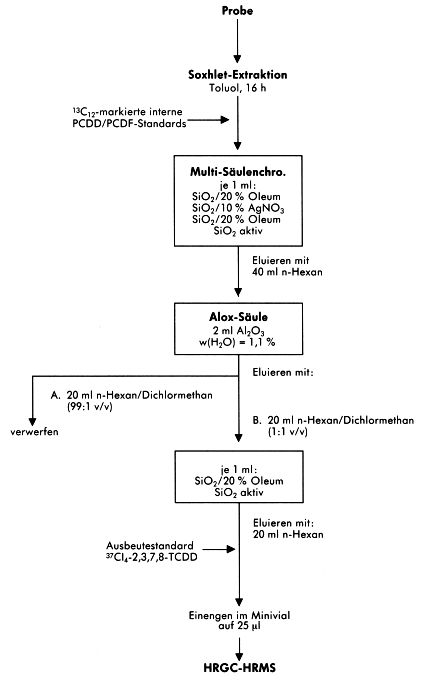
Abbildung 6
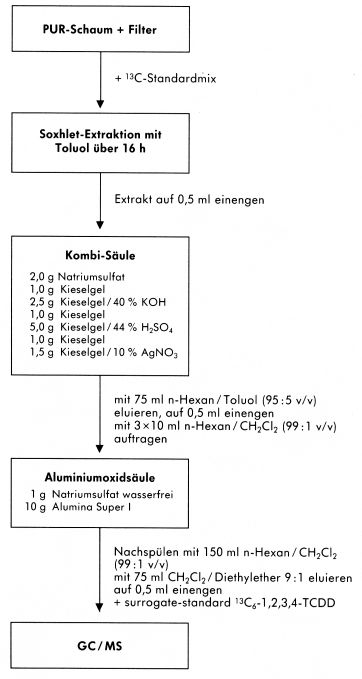
Abbildung 7
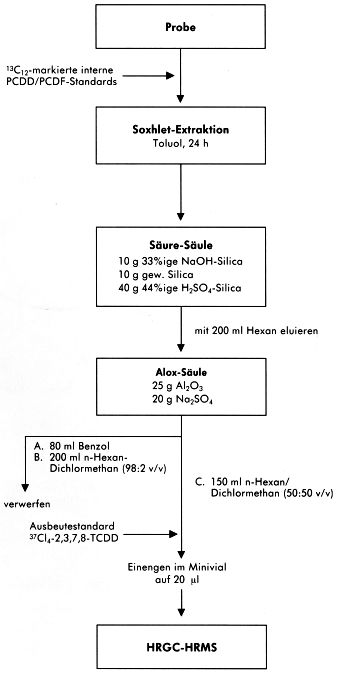
Abbildung 8
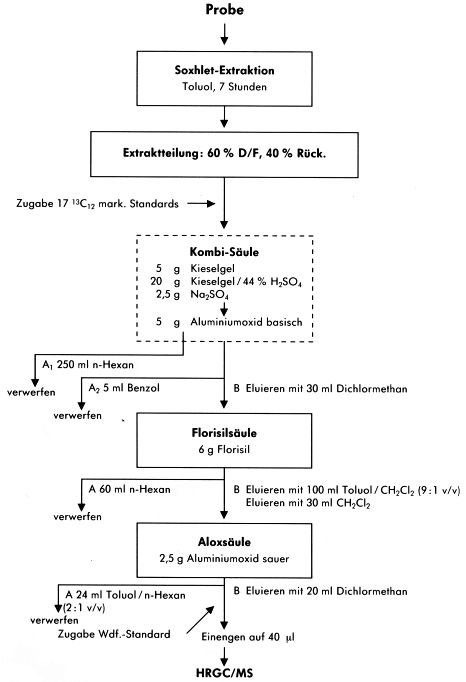
Abbildung 9
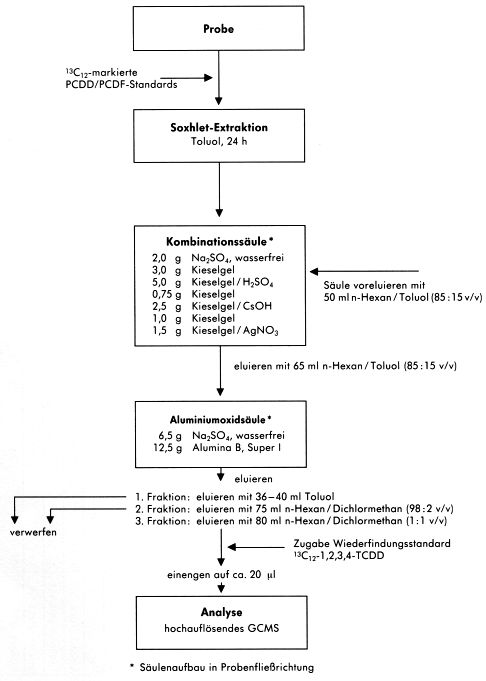
Abbildung 10
Alternative Arbeitsbedingungen für die analytische Bestimmung
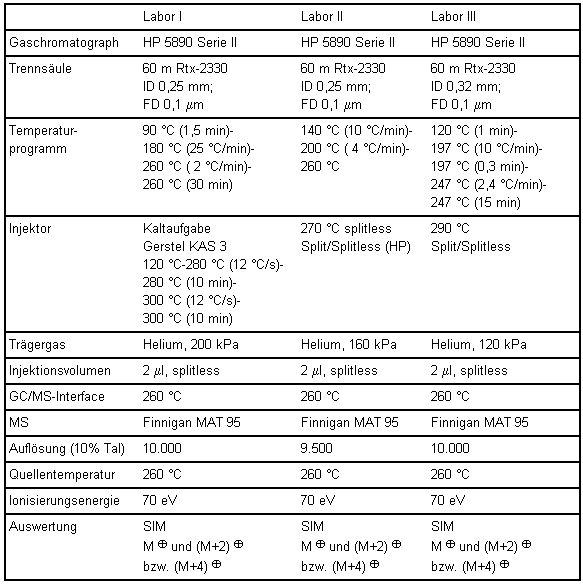
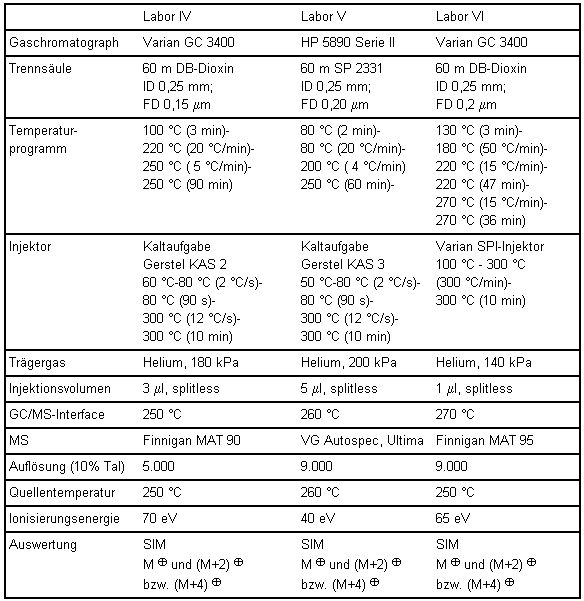
Tabelle 8
__________
1 TDI = Toluylendiisocyanat
2 HRMS = high resolution mass spectrometry
 |
ENDE | |
(Stand: 21.07.2025)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: ab 105.- € netto
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion